Rosalie Griffin Waller, Michael J Madsen, John Gardner, Douglas W Sborov, Nicola J Camp
{"title":"Duo-Shared基因组片段分析确定了骨髓瘤谱系中18q21.33的全基因组显著风险位点。","authors":"Rosalie Griffin Waller, Michael J Madsen, John Gardner, Douglas W Sborov, Nicola J Camp","doi":"10.20517/jtgg.2021.09","DOIUrl":null,"url":null,"abstract":"<p><strong>Aim: </strong>High-risk pedigrees (<i>HRPs</i>) are a powerful design to map highly penetrant risk genes. We previously described Shared Genomic Segment (SGS) analysis, a mapping method for single large extended pedigrees that also addresses genetic heterogeneity inherent in complex diseases. SGS identifies shared segregating chromosomal regions that may inherit in only a subset of cases. However, single large pedigrees that are individually powerful (at least 15 meioses between studied cases) are scarce. Here, we expand the SGS strategy to incorporate evidence from two extended HRPs by identifying the same segregating risk locus in both pedigrees and allowing for some relaxation in the size of each HRP.</p><p><strong>Methods: </strong>Duo-SGS is a procedure to combine single-pedigree SGS evidence. It implements statistically rigorous duo-pedigree thresholding to determine genome-wide significance levels that account for optimization across pedigree pairs. Single-pedigree SGS identifies optimal segments shared by case subsets at each locus across the genome, with nominal significance assessed empirically. Duo-SGS combines the statistical evidence for SGS segments at the same genomic location in two pedigrees using Fisher's method. One pedigree is paired with all others and the best duo-SGS evidence at each locus across the genome is established. Genome-wide significance thresholds are determined through distribution-fitting and the Theory of Large Deviations. We applied the duoSGS strategy to eleven extended, myeloma HRPs.</p><p><strong>Results: </strong>We identified one genome-wide significant region at 18q21.33 (0.85 Mb, <i>P</i> = 7.3 × 10<sup>-9</sup>) which contains one gene, <i>CDH20</i>. Thirteen regions were genome-wide suggestive: 1q42.2, 2p16.1, 3p25.2, 5q21.3, 5q31.1, 6q16.1, 6q26, 7q11.23, 12q24.31, 13q13.3, 18p11.22, 18q22.3 and 19p13.12.</p><p><strong>Conclusion: </strong>Our results provide novel risk loci with segregating evidence from multiple HRPs and offer compelling targets and specific segment carriers to focus a future search for functional variants involved in inherited risk formyeloma.</p>","PeriodicalId":73999,"journal":{"name":"Journal of translational genetics and genomics","volume":"5 2","pages":"112-123"},"PeriodicalIF":1.0000,"publicationDate":"2021-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8654160/pdf/","citationCount":"0","resultStr":"{\"title\":\"Duo Shared Genomic Segment analysis identifies a genome-wide significant risk locus at 18q21.33 in myeloma pedigrees.\",\"authors\":\"Rosalie Griffin Waller, Michael J Madsen, John Gardner, Douglas W Sborov, Nicola J Camp\",\"doi\":\"10.20517/jtgg.2021.09\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Aim: </strong>High-risk pedigrees (<i>HRPs</i>) are a powerful design to map highly penetrant risk genes. We previously described Shared Genomic Segment (SGS) analysis, a mapping method for single large extended pedigrees that also addresses genetic heterogeneity inherent in complex diseases. SGS identifies shared segregating chromosomal regions that may inherit in only a subset of cases. However, single large pedigrees that are individually powerful (at least 15 meioses between studied cases) are scarce. Here, we expand the SGS strategy to incorporate evidence from two extended HRPs by identifying the same segregating risk locus in both pedigrees and allowing for some relaxation in the size of each HRP.</p><p><strong>Methods: </strong>Duo-SGS is a procedure to combine single-pedigree SGS evidence. It implements statistically rigorous duo-pedigree thresholding to determine genome-wide significance levels that account for optimization across pedigree pairs. Single-pedigree SGS identifies optimal segments shared by case subsets at each locus across the genome, with nominal significance assessed empirically. Duo-SGS combines the statistical evidence for SGS segments at the same genomic location in two pedigrees using Fisher's method. One pedigree is paired with all others and the best duo-SGS evidence at each locus across the genome is established. Genome-wide significance thresholds are determined through distribution-fitting and the Theory of Large Deviations. We applied the duoSGS strategy to eleven extended, myeloma HRPs.</p><p><strong>Results: </strong>We identified one genome-wide significant region at 18q21.33 (0.85 Mb, <i>P</i> = 7.3 × 10<sup>-9</sup>) which contains one gene, <i>CDH20</i>. Thirteen regions were genome-wide suggestive: 1q42.2, 2p16.1, 3p25.2, 5q21.3, 5q31.1, 6q16.1, 6q26, 7q11.23, 12q24.31, 13q13.3, 18p11.22, 18q22.3 and 19p13.12.</p><p><strong>Conclusion: </strong>Our results provide novel risk loci with segregating evidence from multiple HRPs and offer compelling targets and specific segment carriers to focus a future search for functional variants involved in inherited risk formyeloma.</p>\",\"PeriodicalId\":73999,\"journal\":{\"name\":\"Journal of translational genetics and genomics\",\"volume\":\"5 2\",\"pages\":\"112-123\"},\"PeriodicalIF\":1.0000,\"publicationDate\":\"2021-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8654160/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of translational genetics and genomics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.20517/jtgg.2021.09\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/5/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of translational genetics and genomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.20517/jtgg.2021.09","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/5/27 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Duo Shared Genomic Segment analysis identifies a genome-wide significant risk locus at 18q21.33 in myeloma pedigrees.
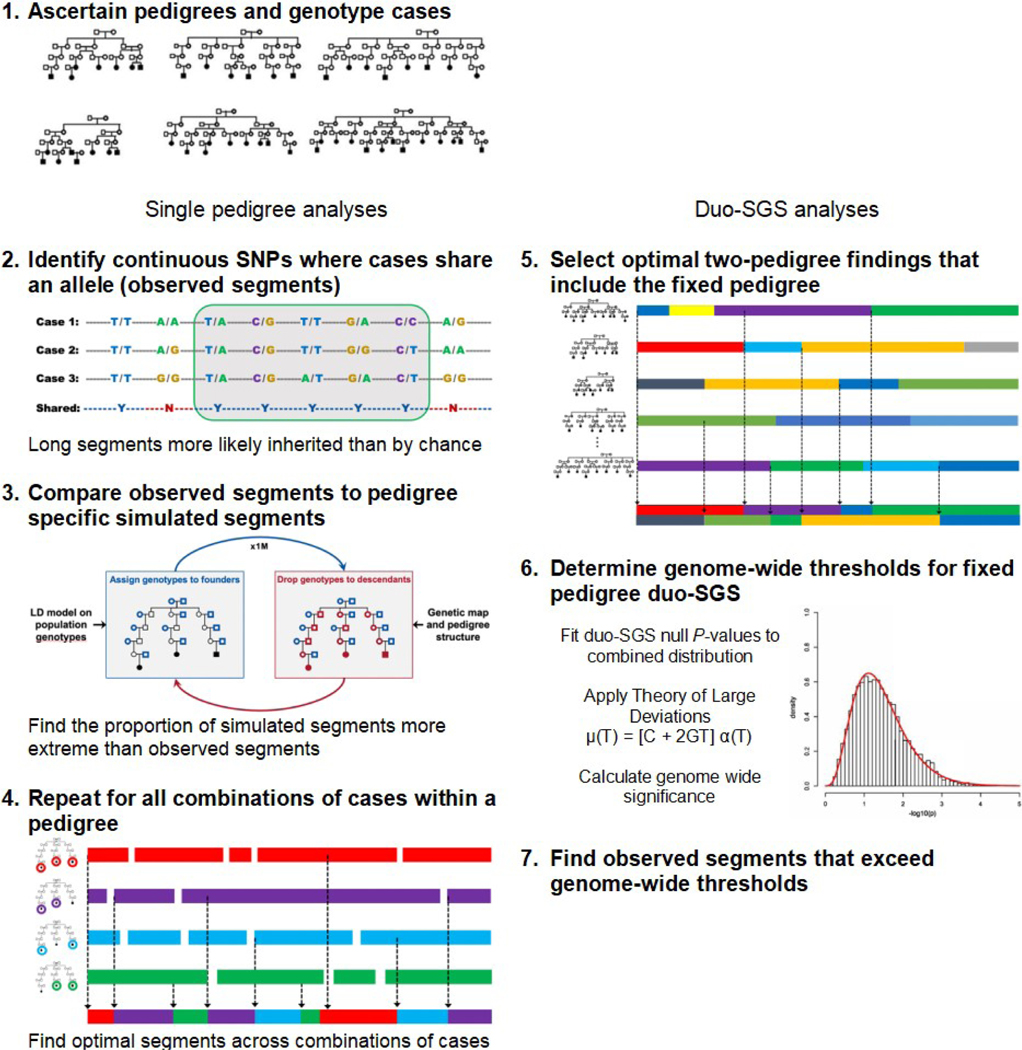
Aim: High-risk pedigrees (HRPs) are a powerful design to map highly penetrant risk genes. We previously described Shared Genomic Segment (SGS) analysis, a mapping method for single large extended pedigrees that also addresses genetic heterogeneity inherent in complex diseases. SGS identifies shared segregating chromosomal regions that may inherit in only a subset of cases. However, single large pedigrees that are individually powerful (at least 15 meioses between studied cases) are scarce. Here, we expand the SGS strategy to incorporate evidence from two extended HRPs by identifying the same segregating risk locus in both pedigrees and allowing for some relaxation in the size of each HRP.
Methods: Duo-SGS is a procedure to combine single-pedigree SGS evidence. It implements statistically rigorous duo-pedigree thresholding to determine genome-wide significance levels that account for optimization across pedigree pairs. Single-pedigree SGS identifies optimal segments shared by case subsets at each locus across the genome, with nominal significance assessed empirically. Duo-SGS combines the statistical evidence for SGS segments at the same genomic location in two pedigrees using Fisher's method. One pedigree is paired with all others and the best duo-SGS evidence at each locus across the genome is established. Genome-wide significance thresholds are determined through distribution-fitting and the Theory of Large Deviations. We applied the duoSGS strategy to eleven extended, myeloma HRPs.
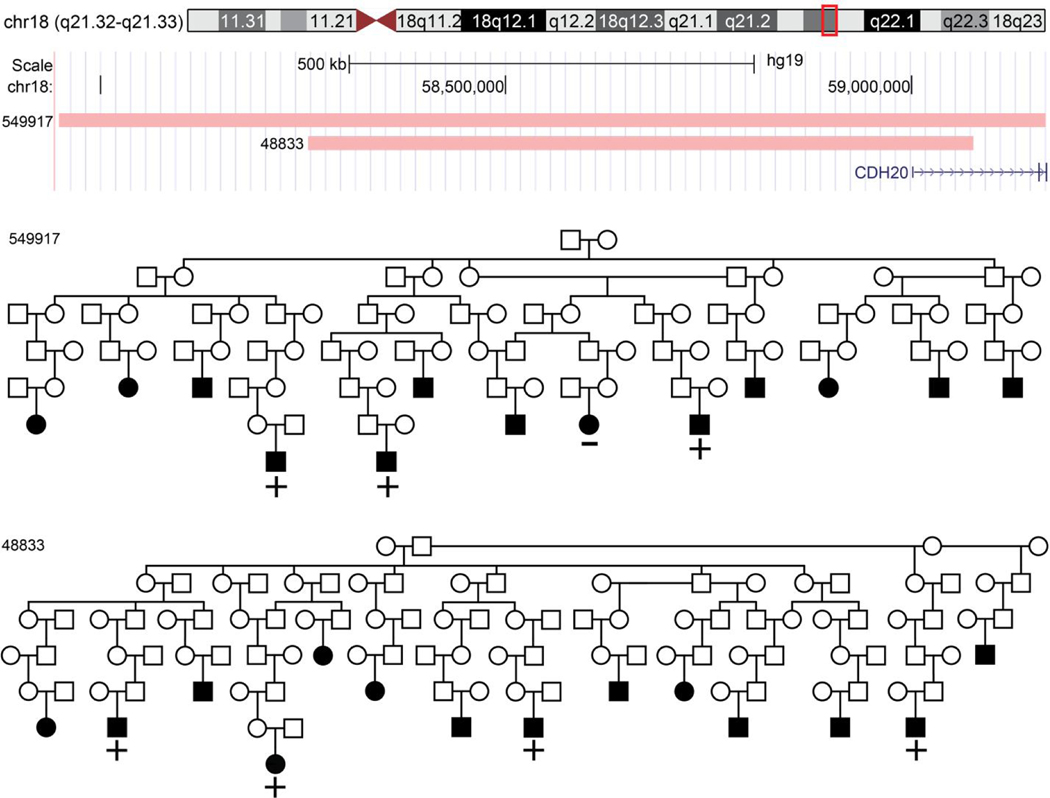
Results: We identified one genome-wide significant region at 18q21.33 (0.85 Mb, P = 7.3 × 10-9) which contains one gene, CDH20. Thirteen regions were genome-wide suggestive: 1q42.2, 2p16.1, 3p25.2, 5q21.3, 5q31.1, 6q16.1, 6q26, 7q11.23, 12q24.31, 13q13.3, 18p11.22, 18q22.3 and 19p13.12.
Conclusion: Our results provide novel risk loci with segregating evidence from multiple HRPs and offer compelling targets and specific segment carriers to focus a future search for functional variants involved in inherited risk formyeloma.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


