Robert J O'Shea, Sophia Tsoka, Gary Jr Cook, Vicky Goh
{"title":"癌症基因组学中的稀疏回归:比较真实世界数据中的变量选择和预测。","authors":"Robert J O'Shea, Sophia Tsoka, Gary Jr Cook, Vicky Goh","doi":"10.1177/11769351211056298","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Evaluation of gene interaction models in cancer genomics is challenging, as the true distribution is uncertain. Previous analyses have benchmarked models using synthetic data or databases of experimentally verified interactions - approaches which are susceptible to misrepresentation and incompleteness, respectively. The objectives of this analysis are to (1) provide a real-world data-driven approach for comparing performance of genomic model inference algorithms, (2) compare the performance of LASSO, elastic net, best-subset selection, <math> <mrow><msub><mi>L</mi> <mrow><mn>0</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>1</mn></mrow> </msub> </mrow> </math> penalisation and <math> <mrow><msub><mi>L</mi> <mrow><mn>0</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> penalisation in real genomic data and (3) compare algorithmic preselection according to performance in our benchmark datasets to algorithmic selection by internal cross-validation.</p><p><strong>Methods: </strong>Five large <math><mrow><mo>(</mo> <mi>n</mi> <mn>4000</mn> <mo>)</mo></mrow> </math> genomic datasets were extracted from Gene Expression Omnibus. 'Gold-standard' regression models were trained on subspaces of these datasets ( <math><mrow><mi>n</mi> <mn>4000</mn></mrow> </math> , <math><mrow><mi>p</mi> <mo>=</mo> <mn>500</mn></mrow> </math> ). Penalised regression models were trained on small samples from these subspaces ( <math><mrow><mi>n</mi> <mo>∈</mo> <mrow><mo>{</mo> <mrow><mn>25</mn> <mo>,</mo> <mn>75</mn> <mo>,</mo> <mn>150</mn></mrow> <mo>}</mo></mrow> <mo>,</mo> <mi>p</mi> <mo>=</mo> <mn>500</mn></mrow> </math> ) and validated against the gold-standard models. Variable selection performance and out-of-sample prediction were assessed. Penalty 'preselection' according to test performance in the other 4 datasets was compared to selection internal cross-validation error minimisation.</p><p><strong>Results: </strong><math> <mrow><msub><mi>L</mi> <mrow><mn>1</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> -penalisation achieved the highest cosine similarity between estimated coefficients and those of gold-standard models. <math> <mrow><msub><mi>L</mi> <mrow><mn>0</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> -penalised models explained the greatest proportion of variance in test responses, though performance was unreliable in low signal:noise conditions. <math> <mrow><msub><mi>L</mi> <mrow><mn>0</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> also attained the highest overall median variable selection F1 score. Penalty preselection significantly outperformed selection by internal cross-validation in each of 3 examined metrics.</p><p><strong>Conclusions: </strong>This analysis explores a novel approach for comparisons of model selection approaches in real genomic data from 5 cancers. Our benchmarking datasets have been made publicly available for use in future research. Our findings support the use of <math> <mrow><msub><mi>L</mi> <mrow><mn>0</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> penalisation for structural selection and <math> <mrow><msub><mi>L</mi> <mrow><mn>1</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> penalisation for coefficient recovery in genomic data. Evaluation of learning algorithms according to observed test performance in external genomic datasets yields valuable insights into actual test performance, providing a data-driven complement to internal cross-validation in genomic regression tasks.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":"20 ","pages":"11769351211056298"},"PeriodicalIF":2.4000,"publicationDate":"2021-11-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8640984/pdf/","citationCount":"0","resultStr":"{\"title\":\"Sparse Regression in Cancer Genomics: Comparing Variable Selection and Predictions in Real World Data.\",\"authors\":\"Robert J O'Shea, Sophia Tsoka, Gary Jr Cook, Vicky Goh\",\"doi\":\"10.1177/11769351211056298\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Evaluation of gene interaction models in cancer genomics is challenging, as the true distribution is uncertain. Previous analyses have benchmarked models using synthetic data or databases of experimentally verified interactions - approaches which are susceptible to misrepresentation and incompleteness, respectively. The objectives of this analysis are to (1) provide a real-world data-driven approach for comparing performance of genomic model inference algorithms, (2) compare the performance of LASSO, elastic net, best-subset selection, <math> <mrow><msub><mi>L</mi> <mrow><mn>0</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>1</mn></mrow> </msub> </mrow> </math> penalisation and <math> <mrow><msub><mi>L</mi> <mrow><mn>0</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> penalisation in real genomic data and (3) compare algorithmic preselection according to performance in our benchmark datasets to algorithmic selection by internal cross-validation.</p><p><strong>Methods: </strong>Five large <math><mrow><mo>(</mo> <mi>n</mi> <mn>4000</mn> <mo>)</mo></mrow> </math> genomic datasets were extracted from Gene Expression Omnibus. 'Gold-standard' regression models were trained on subspaces of these datasets ( <math><mrow><mi>n</mi> <mn>4000</mn></mrow> </math> , <math><mrow><mi>p</mi> <mo>=</mo> <mn>500</mn></mrow> </math> ). Penalised regression models were trained on small samples from these subspaces ( <math><mrow><mi>n</mi> <mo>∈</mo> <mrow><mo>{</mo> <mrow><mn>25</mn> <mo>,</mo> <mn>75</mn> <mo>,</mo> <mn>150</mn></mrow> <mo>}</mo></mrow> <mo>,</mo> <mi>p</mi> <mo>=</mo> <mn>500</mn></mrow> </math> ) and validated against the gold-standard models. Variable selection performance and out-of-sample prediction were assessed. Penalty 'preselection' according to test performance in the other 4 datasets was compared to selection internal cross-validation error minimisation.</p><p><strong>Results: </strong><math> <mrow><msub><mi>L</mi> <mrow><mn>1</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> -penalisation achieved the highest cosine similarity between estimated coefficients and those of gold-standard models. <math> <mrow><msub><mi>L</mi> <mrow><mn>0</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> -penalised models explained the greatest proportion of variance in test responses, though performance was unreliable in low signal:noise conditions. <math> <mrow><msub><mi>L</mi> <mrow><mn>0</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> also attained the highest overall median variable selection F1 score. Penalty preselection significantly outperformed selection by internal cross-validation in each of 3 examined metrics.</p><p><strong>Conclusions: </strong>This analysis explores a novel approach for comparisons of model selection approaches in real genomic data from 5 cancers. Our benchmarking datasets have been made publicly available for use in future research. Our findings support the use of <math> <mrow><msub><mi>L</mi> <mrow><mn>0</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> penalisation for structural selection and <math> <mrow><msub><mi>L</mi> <mrow><mn>1</mn></mrow> </msub> <msub><mi>L</mi> <mrow><mn>2</mn></mrow> </msub> </mrow> </math> penalisation for coefficient recovery in genomic data. Evaluation of learning algorithms according to observed test performance in external genomic datasets yields valuable insights into actual test performance, providing a data-driven complement to internal cross-validation in genomic regression tasks.</p>\",\"PeriodicalId\":35418,\"journal\":{\"name\":\"Cancer Informatics\",\"volume\":\"20 \",\"pages\":\"11769351211056298\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2021-11-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8640984/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cancer Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11769351211056298\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351211056298","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
背景:评估癌症基因组学中的基因相互作用模型具有挑战性,因为真实的分布是不确定的。以往的分析都是使用合成数据或实验验证的相互作用数据库对模型进行基准测试,而这两种方法分别容易受到错误表述和不完整性的影响。本分析的目的是:(1) 提供一种真实世界数据驱动的方法,用于比较基因组模型推断算法的性能;(2) 比较 LASSO、弹性网、最佳子集选择、L 0 L 1 惩罚和 L 0 L 2 惩罚在真实基因组数据中的性能;(3) 比较根据基准数据集性能进行的算法预选和通过内部交叉验证进行的算法选择:从基因表达总库(Gene Expression Omnibus)中提取了五个大型(n 4000)基因组数据集。黄金标准 "回归模型在这些数据集的子空间(n 4000,p = 500)上进行训练。在这些子空间的小样本(n ∈ { 25 , 75 , 150 } , p = 500)上训练惩罚回归模型,并与黄金标准模型进行验证。对变量选择性能和样本外预测进行了评估。根据其他 4 个数据集的测试结果进行的惩罚性 "预选 "与内部交叉验证误差最小化的选择进行了比较: L 1 L 2 惩罚法在估计系数和黄金标准模型系数之间取得了最高的余弦相似度。 L 0 L 2 放大模型解释了测试反应中最大比例的变异,尽管在低信噪比条件下表现并不可靠。 L 0 L 2 还获得了最高的变量选择 F1 总分中值。在 3 个考察指标中,惩罚预选在每个指标上都明显优于内部交叉验证选择:这项分析探索了一种新方法,用于在 5 种癌症的真实基因组数据中比较模型选择方法。我们的基准数据集已公开发布,供未来研究使用。我们的研究结果支持在基因组数据中使用 L 0 L 2 惩罚进行结构选择,使用 L 1 L 2 惩罚进行系数恢复。根据在外部基因组数据集中观察到的测试性能来评估学习算法,可以获得对实际测试性能的宝贵见解,为基因组回归任务中的内部交叉验证提供了数据驱动的补充。
Sparse Regression in Cancer Genomics: Comparing Variable Selection and Predictions in Real World Data.
Background: Evaluation of gene interaction models in cancer genomics is challenging, as the true distribution is uncertain. Previous analyses have benchmarked models using synthetic data or databases of experimentally verified interactions - approaches which are susceptible to misrepresentation and incompleteness, respectively. The objectives of this analysis are to (1) provide a real-world data-driven approach for comparing performance of genomic model inference algorithms, (2) compare the performance of LASSO, elastic net, best-subset selection, penalisation and penalisation in real genomic data and (3) compare algorithmic preselection according to performance in our benchmark datasets to algorithmic selection by internal cross-validation.
Methods: Five large genomic datasets were extracted from Gene Expression Omnibus. 'Gold-standard' regression models were trained on subspaces of these datasets ( , ). Penalised regression models were trained on small samples from these subspaces ( ) and validated against the gold-standard models. Variable selection performance and out-of-sample prediction were assessed. Penalty 'preselection' according to test performance in the other 4 datasets was compared to selection internal cross-validation error minimisation.
Results: -penalisation achieved the highest cosine similarity between estimated coefficients and those of gold-standard models. -penalised models explained the greatest proportion of variance in test responses, though performance was unreliable in low signal:noise conditions. also attained the highest overall median variable selection F1 score. Penalty preselection significantly outperformed selection by internal cross-validation in each of 3 examined metrics.
Conclusions: This analysis explores a novel approach for comparisons of model selection approaches in real genomic data from 5 cancers. Our benchmarking datasets have been made publicly available for use in future research. Our findings support the use of penalisation for structural selection and penalisation for coefficient recovery in genomic data. Evaluation of learning algorithms according to observed test performance in external genomic datasets yields valuable insights into actual test performance, providing a data-driven complement to internal cross-validation in genomic regression tasks.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.

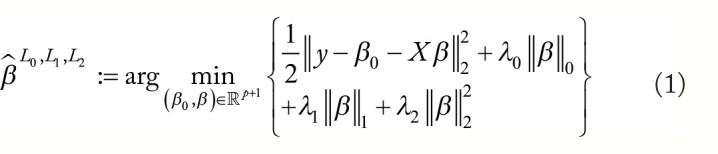

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


