{"title":"比较良性和恶性乳腺肿瘤患者的肠道微生物群:一项试点研究","authors":"Peidong Yang, Zhitang Wang, Qingqin Peng, Weibin Lian, Debo Chen","doi":"10.1177/11769343211057573","DOIUrl":null,"url":null,"abstract":"<p><p>The microbiome plays diverse roles in many diseases and can potentially contribute to cancer development. Breast cancer is the most commonly diagnosed cancer in women worldwide. Thus, we investigated whether the gut microbiota differs between patients with breast carcinoma and those with benign tumors. The DNA of the fecal microbiota community was detected by Illumina sequencing and the taxonomy of 16S rRNA genes. The α-diversity and β-diversity analyses were used to determine richness and evenness of the gut microbiota. Gene function prediction of the microbiota in patients with benign and malignant carcinoma was performed using PICRUSt. There was no significant difference in the α-diversity between patients with benign and malignant tumors (<i>P</i> = 3.15e<sup>-1</sup> for the Chao index and <i>P</i> = 3.1e<sup>-1</sup> for the ACE index). The microbiota composition was different between the 2 groups, although no statistical difference was observed in β-diversity. Of the 31 different genera compared between the 2 groups, level of only <i>Citrobacter</i> was significantly higher in the malignant tumor group than that in benign tumor group. The metabolic pathways of the gut microbiome in the malignant tumor group were significantly different from those in benign tumor group. Furthermore, the study establishes the distinct richness of the gut microbiome in patients with breast cancer with different clinicopathological factors, including ER, PR, Ki-67 level, Her2 status, and tumor grade. These findings suggest that the gut microbiome may be useful for the diagnosis and treatment of malignant breast carcinoma.</p>","PeriodicalId":50472,"journal":{"name":"Evolutionary Bioinformatics","volume":"17 ","pages":"11769343211057573"},"PeriodicalIF":1.7000,"publicationDate":"2021-11-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/b1/42/10.1177_11769343211057573.PMC8593289.pdf","citationCount":"0","resultStr":"{\"title\":\"Comparison of the Gut Microbiota in Patients with Benign and Malignant Breast Tumors: A Pilot Study.\",\"authors\":\"Peidong Yang, Zhitang Wang, Qingqin Peng, Weibin Lian, Debo Chen\",\"doi\":\"10.1177/11769343211057573\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The microbiome plays diverse roles in many diseases and can potentially contribute to cancer development. Breast cancer is the most commonly diagnosed cancer in women worldwide. Thus, we investigated whether the gut microbiota differs between patients with breast carcinoma and those with benign tumors. The DNA of the fecal microbiota community was detected by Illumina sequencing and the taxonomy of 16S rRNA genes. The α-diversity and β-diversity analyses were used to determine richness and evenness of the gut microbiota. Gene function prediction of the microbiota in patients with benign and malignant carcinoma was performed using PICRUSt. There was no significant difference in the α-diversity between patients with benign and malignant tumors (<i>P</i> = 3.15e<sup>-1</sup> for the Chao index and <i>P</i> = 3.1e<sup>-1</sup> for the ACE index). The microbiota composition was different between the 2 groups, although no statistical difference was observed in β-diversity. Of the 31 different genera compared between the 2 groups, level of only <i>Citrobacter</i> was significantly higher in the malignant tumor group than that in benign tumor group. The metabolic pathways of the gut microbiome in the malignant tumor group were significantly different from those in benign tumor group. Furthermore, the study establishes the distinct richness of the gut microbiome in patients with breast cancer with different clinicopathological factors, including ER, PR, Ki-67 level, Her2 status, and tumor grade. These findings suggest that the gut microbiome may be useful for the diagnosis and treatment of malignant breast carcinoma.</p>\",\"PeriodicalId\":50472,\"journal\":{\"name\":\"Evolutionary Bioinformatics\",\"volume\":\"17 \",\"pages\":\"11769343211057573\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2021-11-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/b1/42/10.1177_11769343211057573.PMC8593289.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Evolutionary Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1177/11769343211057573\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"EVOLUTIONARY BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Evolutionary Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1177/11769343211057573","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
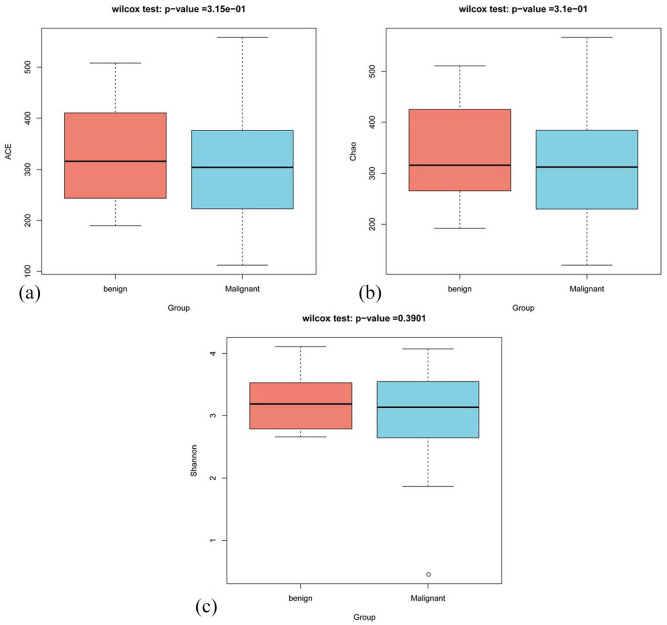
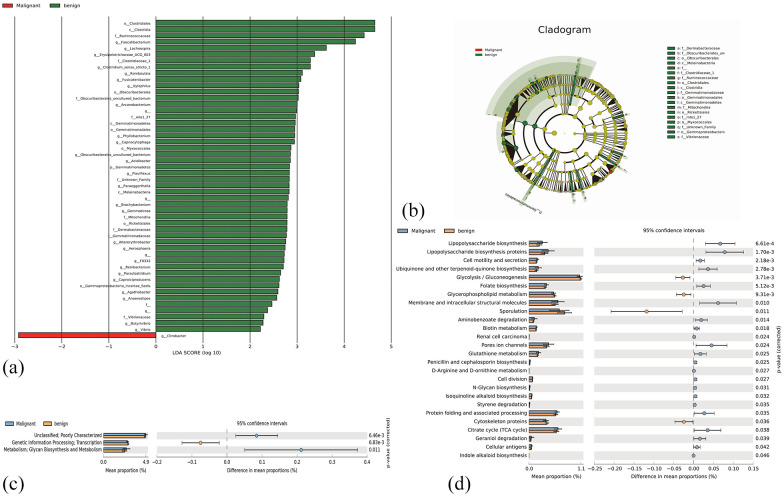
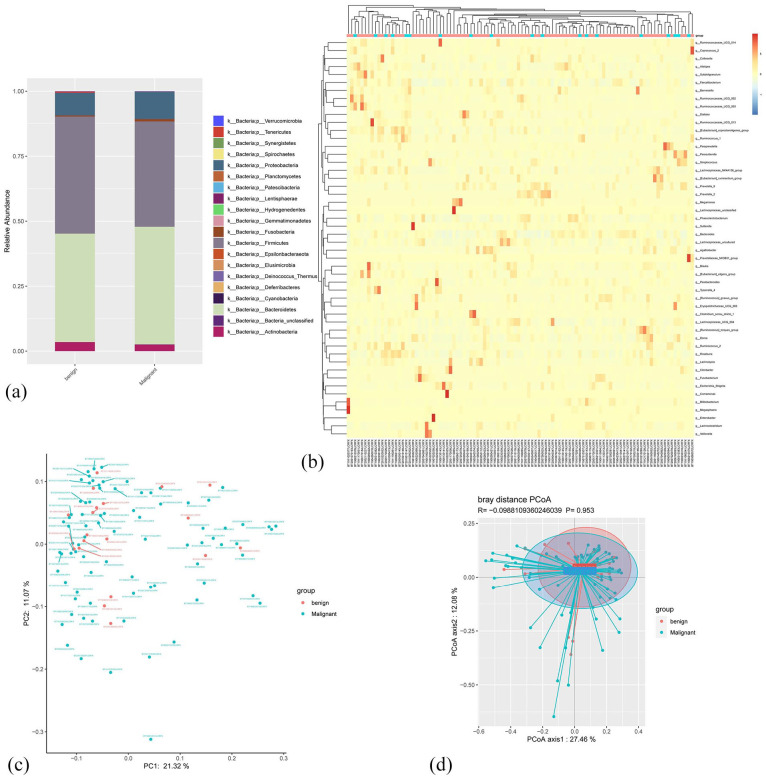
微生物组在许多疾病中发挥着不同的作用,并有可能导致癌症的发生。乳腺癌是全球妇女最常确诊的癌症。因此,我们研究了乳腺癌患者和良性肿瘤患者的肠道微生物群是否存在差异。我们通过 Illumina 测序和 16S rRNA 基因分类检测了粪便微生物群落的 DNA。α多样性和β多样性分析用于确定肠道微生物群的丰富度和均匀度。使用 PICRUSt 对良性和恶性肿瘤患者的微生物群进行了基因功能预测,结果发现良性和恶性肿瘤患者的 α 多样性无显著差异(Chao 指数为 P = 3.15e-1,ACE 指数为 P = 3.1e-1)。两组患者的微生物群组成不同,但在β多样性方面未观察到统计学差异。在两组比较的31个不同菌属中,恶性肿瘤组中只有柠檬酸杆菌的水平明显高于良性肿瘤组。恶性肿瘤组肠道微生物组的代谢途径与良性肿瘤组明显不同。此外,该研究还确定了不同临床病理因素(包括ER、PR、Ki-67水平、Her2状态和肿瘤分级)的乳腺癌患者肠道微生物组的不同丰富程度。这些发现表明,肠道微生物组可能有助于恶性乳腺癌的诊断和治疗。
Comparison of the Gut Microbiota in Patients with Benign and Malignant Breast Tumors: A Pilot Study.
The microbiome plays diverse roles in many diseases and can potentially contribute to cancer development. Breast cancer is the most commonly diagnosed cancer in women worldwide. Thus, we investigated whether the gut microbiota differs between patients with breast carcinoma and those with benign tumors. The DNA of the fecal microbiota community was detected by Illumina sequencing and the taxonomy of 16S rRNA genes. The α-diversity and β-diversity analyses were used to determine richness and evenness of the gut microbiota. Gene function prediction of the microbiota in patients with benign and malignant carcinoma was performed using PICRUSt. There was no significant difference in the α-diversity between patients with benign and malignant tumors (P = 3.15e-1 for the Chao index and P = 3.1e-1 for the ACE index). The microbiota composition was different between the 2 groups, although no statistical difference was observed in β-diversity. Of the 31 different genera compared between the 2 groups, level of only Citrobacter was significantly higher in the malignant tumor group than that in benign tumor group. The metabolic pathways of the gut microbiome in the malignant tumor group were significantly different from those in benign tumor group. Furthermore, the study establishes the distinct richness of the gut microbiome in patients with breast cancer with different clinicopathological factors, including ER, PR, Ki-67 level, Her2 status, and tumor grade. These findings suggest that the gut microbiome may be useful for the diagnosis and treatment of malignant breast carcinoma.
期刊介绍:
Evolutionary Bioinformatics is an open access, peer reviewed international journal focusing on evolutionary bioinformatics. The journal aims to support understanding of organismal form and function through use of molecular, genetic, genomic and proteomic data by giving due consideration to its evolutionary context.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


