Jie Liu, Yuanhao Huang, Ritambhara Singh, Jean-Philippe Vert, William Stafford Noble
{"title":"联合嵌入多个单细胞组学测量。","authors":"Jie Liu, Yuanhao Huang, Ritambhara Singh, Jean-Philippe Vert, William Stafford Noble","doi":"10.4230/LIPIcs.WABI.2019.10","DOIUrl":null,"url":null,"abstract":"<p><p>Many single-cell sequencing technologies are now available, but it is still difficult to apply multiple sequencing technologies to the same single cell. In this paper, we propose an unsupervised manifold alignment algorithm, MMD-MA, for integrating multiple measurements carried out on disjoint aliquots of a given population of cells. Effectively, MMD-MA performs an <i>in silico</i> co-assay by embedding cells measured in different ways into a learned latent space. In the MMD-MA algorithm, single-cell data points from multiple domains are aligned by optimizing an objective function with three components: (1) a maximum mean discrepancy (MMD) term to encourage the differently measured points to have similar distributions in the latent space, (2) a distortion term to preserve the structure of the data between the input space and the latent space, and (3) a penalty term to avoid collapse to a trivial solution. Notably, MMD-MA does not require any correspondence information across data modalities, either between the cells or between the features. Furthermore, MMD-MA's weak distributional requirements for the domains to be aligned allow the algorithm to integrate heterogeneous types of single cell measures, such as gene expression, DNA accessibility, chromatin organization, methylation, and imaging data. We demonstrate the utility of MMD-MA in simulation experiments and using a real data set involving single-cell gene expression and methylation data.</p>","PeriodicalId":93254,"journal":{"name":"Algorithms in bioinformatics : ... International Workshop, WABI ..., proceedings. WABI (Workshop)","volume":"143 ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2019-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8496402/pdf/","citationCount":"0","resultStr":"{\"title\":\"Jointly Embedding Multiple Single-Cell Omics Measurements.\",\"authors\":\"Jie Liu, Yuanhao Huang, Ritambhara Singh, Jean-Philippe Vert, William Stafford Noble\",\"doi\":\"10.4230/LIPIcs.WABI.2019.10\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Many single-cell sequencing technologies are now available, but it is still difficult to apply multiple sequencing technologies to the same single cell. In this paper, we propose an unsupervised manifold alignment algorithm, MMD-MA, for integrating multiple measurements carried out on disjoint aliquots of a given population of cells. Effectively, MMD-MA performs an <i>in silico</i> co-assay by embedding cells measured in different ways into a learned latent space. In the MMD-MA algorithm, single-cell data points from multiple domains are aligned by optimizing an objective function with three components: (1) a maximum mean discrepancy (MMD) term to encourage the differently measured points to have similar distributions in the latent space, (2) a distortion term to preserve the structure of the data between the input space and the latent space, and (3) a penalty term to avoid collapse to a trivial solution. Notably, MMD-MA does not require any correspondence information across data modalities, either between the cells or between the features. Furthermore, MMD-MA's weak distributional requirements for the domains to be aligned allow the algorithm to integrate heterogeneous types of single cell measures, such as gene expression, DNA accessibility, chromatin organization, methylation, and imaging data. We demonstrate the utility of MMD-MA in simulation experiments and using a real data set involving single-cell gene expression and methylation data.</p>\",\"PeriodicalId\":93254,\"journal\":{\"name\":\"Algorithms in bioinformatics : ... International Workshop, WABI ..., proceedings. WABI (Workshop)\",\"volume\":\"143 \",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2019-09-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8496402/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Algorithms in bioinformatics : ... International Workshop, WABI ..., proceedings. WABI (Workshop)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.4230/LIPIcs.WABI.2019.10\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms in bioinformatics : ... International Workshop, WABI ..., proceedings. WABI (Workshop)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4230/LIPIcs.WABI.2019.10","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
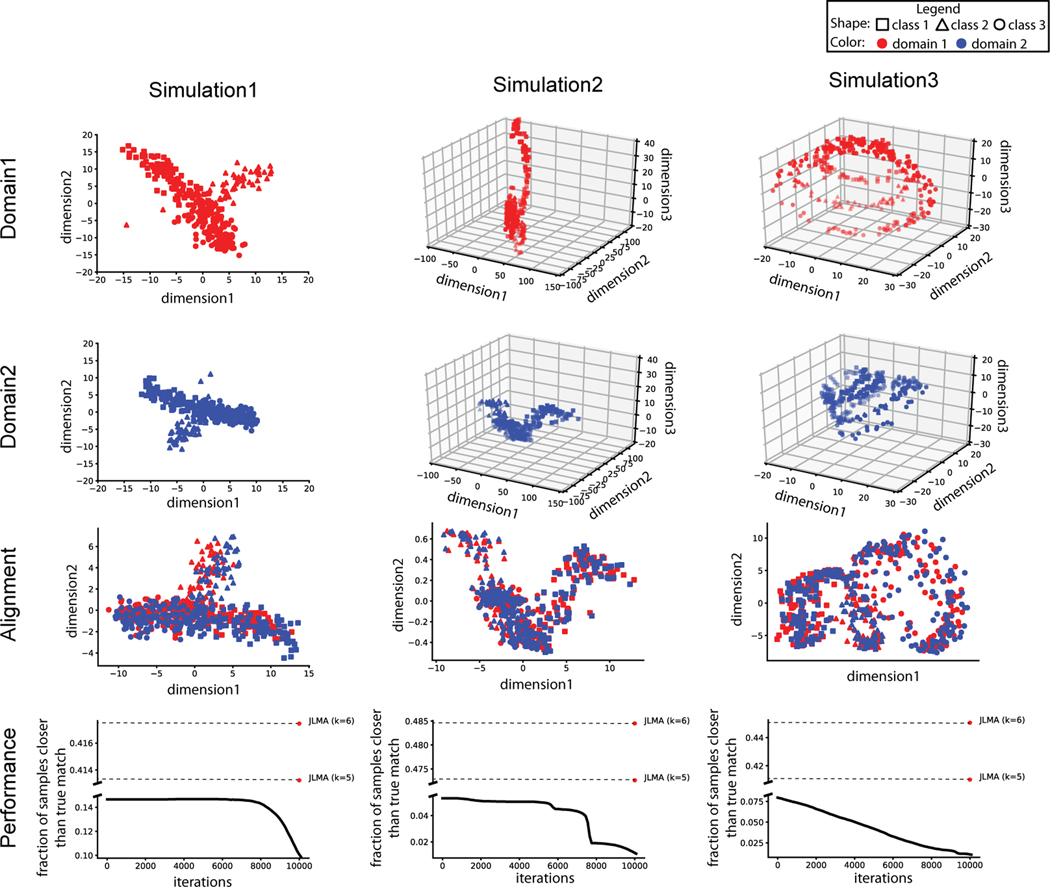
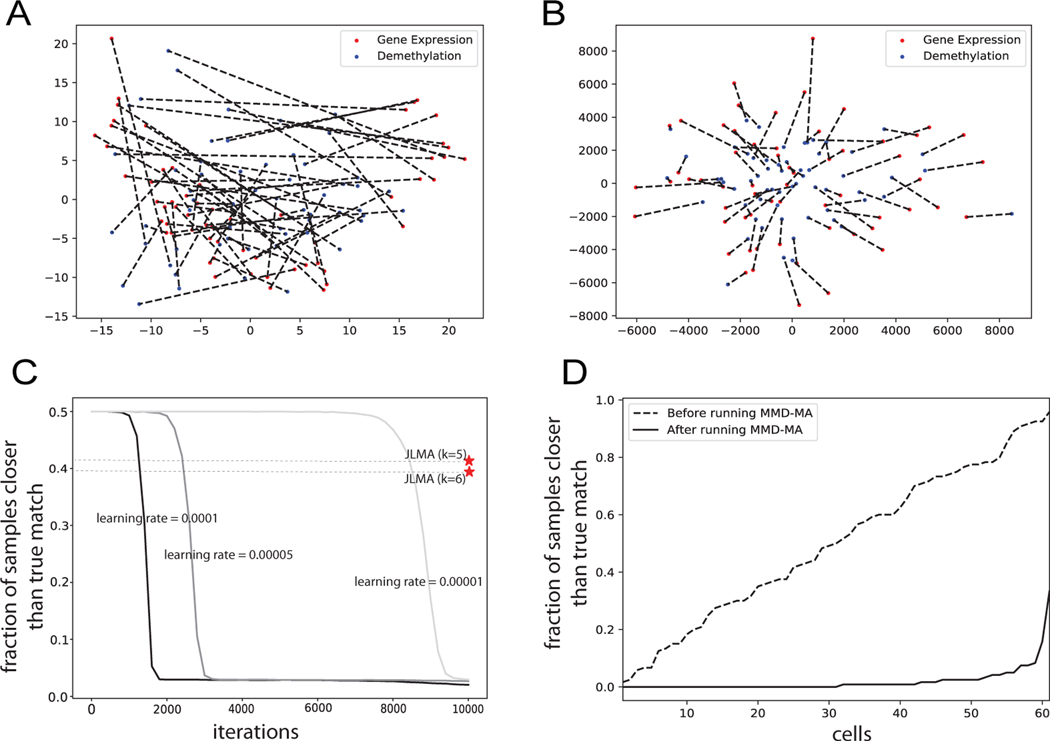
Many single-cell sequencing technologies are now available, but it is still difficult to apply multiple sequencing technologies to the same single cell. In this paper, we propose an unsupervised manifold alignment algorithm, MMD-MA, for integrating multiple measurements carried out on disjoint aliquots of a given population of cells. Effectively, MMD-MA performs an in silico co-assay by embedding cells measured in different ways into a learned latent space. In the MMD-MA algorithm, single-cell data points from multiple domains are aligned by optimizing an objective function with three components: (1) a maximum mean discrepancy (MMD) term to encourage the differently measured points to have similar distributions in the latent space, (2) a distortion term to preserve the structure of the data between the input space and the latent space, and (3) a penalty term to avoid collapse to a trivial solution. Notably, MMD-MA does not require any correspondence information across data modalities, either between the cells or between the features. Furthermore, MMD-MA's weak distributional requirements for the domains to be aligned allow the algorithm to integrate heterogeneous types of single cell measures, such as gene expression, DNA accessibility, chromatin organization, methylation, and imaging data. We demonstrate the utility of MMD-MA in simulation experiments and using a real data set involving single-cell gene expression and methylation data.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


