Paul Little, Heejoon Jo, Alan Hoyle, Angela Mazul, Xiaobei Zhao, Ashley H Salazar, Douglas Farquhar, Siddharth Sheth, Maheer Masood, Michele C Hayward, Joel S Parker, Katherine A Hoadley, Jose Zevallos, D Neil Hayes
{"title":"UNMASC:只有肿瘤的变种呼叫与不匹配的正常对照。","authors":"Paul Little, Heejoon Jo, Alan Hoyle, Angela Mazul, Xiaobei Zhao, Ashley H Salazar, Douglas Farquhar, Siddharth Sheth, Maheer Masood, Michele C Hayward, Joel S Parker, Katherine A Hoadley, Jose Zevallos, D Neil Hayes","doi":"10.1093/narcan/zcab040","DOIUrl":null,"url":null,"abstract":"<p><p>Despite years of progress, mutation detection in cancer samples continues to require significant manual review as a final step. Expert review is particularly challenging in cases where tumors are sequenced without matched normal control DNA. Attempts have been made to call somatic point mutations without a matched normal sample by removing well-known germline variants, utilizing unmatched normal controls, and constructing decision rules to classify sequencing errors and private germline variants. With budgetary constraints related to computational and sequencing costs, finding the appropriate number of controls is a crucial step to identifying somatic variants. Our approach utilizes public databases for canonical somatic variants as well as germline variants and leverages information gathered about nearby positions in the normal controls. Drawing from our cohort of targeted capture panel sequencing of tumor and normal samples with varying tumortypes and demographics, these served as a benchmark for our tumor-only variant calling pipeline to observe the relationship between our ability to correctly classify variants against a number of unmatched normals. With our benchmarked samples, approximately ten normal controls were needed to maintain 94% sensitivity, 99% specificity and 76% positive predictive value, far outperforming comparable methods. Our approach, called UNMASC, also serves as a supplement to traditional tumor with matched normal variant calling workflows and can potentially extend to other concerns arising from analyzing next generation sequencing data.</p>","PeriodicalId":18879,"journal":{"name":"NAR Cancer","volume":" ","pages":"zcab040"},"PeriodicalIF":0.0000,"publicationDate":"2021-10-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8494212/pdf/","citationCount":"4","resultStr":"{\"title\":\"UNMASC: tumor-only variant calling with unmatched normal controls.\",\"authors\":\"Paul Little, Heejoon Jo, Alan Hoyle, Angela Mazul, Xiaobei Zhao, Ashley H Salazar, Douglas Farquhar, Siddharth Sheth, Maheer Masood, Michele C Hayward, Joel S Parker, Katherine A Hoadley, Jose Zevallos, D Neil Hayes\",\"doi\":\"10.1093/narcan/zcab040\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Despite years of progress, mutation detection in cancer samples continues to require significant manual review as a final step. Expert review is particularly challenging in cases where tumors are sequenced without matched normal control DNA. Attempts have been made to call somatic point mutations without a matched normal sample by removing well-known germline variants, utilizing unmatched normal controls, and constructing decision rules to classify sequencing errors and private germline variants. With budgetary constraints related to computational and sequencing costs, finding the appropriate number of controls is a crucial step to identifying somatic variants. Our approach utilizes public databases for canonical somatic variants as well as germline variants and leverages information gathered about nearby positions in the normal controls. Drawing from our cohort of targeted capture panel sequencing of tumor and normal samples with varying tumortypes and demographics, these served as a benchmark for our tumor-only variant calling pipeline to observe the relationship between our ability to correctly classify variants against a number of unmatched normals. With our benchmarked samples, approximately ten normal controls were needed to maintain 94% sensitivity, 99% specificity and 76% positive predictive value, far outperforming comparable methods. Our approach, called UNMASC, also serves as a supplement to traditional tumor with matched normal variant calling workflows and can potentially extend to other concerns arising from analyzing next generation sequencing data.</p>\",\"PeriodicalId\":18879,\"journal\":{\"name\":\"NAR Cancer\",\"volume\":\" \",\"pages\":\"zcab040\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-10-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8494212/pdf/\",\"citationCount\":\"4\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NAR Cancer\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/narcan/zcab040\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/12/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Cancer","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/narcan/zcab040","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/12/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
UNMASC: tumor-only variant calling with unmatched normal controls.
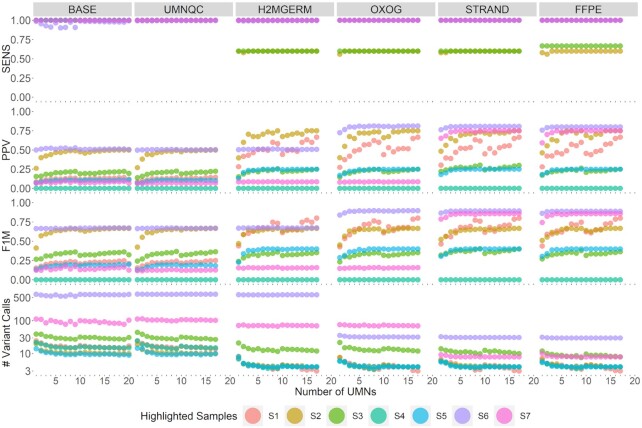
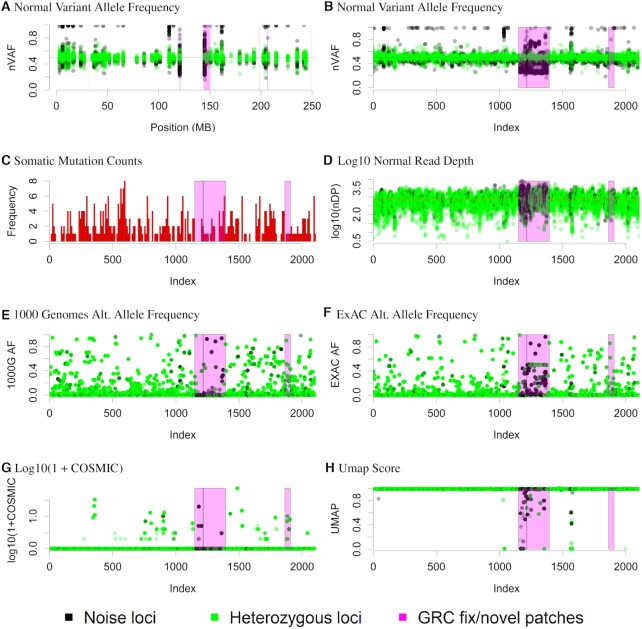
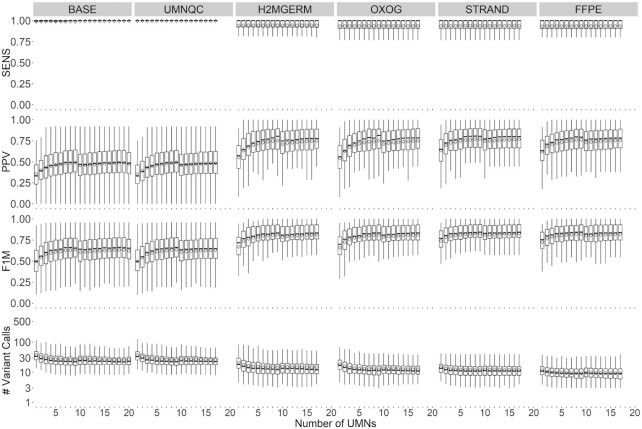
Despite years of progress, mutation detection in cancer samples continues to require significant manual review as a final step. Expert review is particularly challenging in cases where tumors are sequenced without matched normal control DNA. Attempts have been made to call somatic point mutations without a matched normal sample by removing well-known germline variants, utilizing unmatched normal controls, and constructing decision rules to classify sequencing errors and private germline variants. With budgetary constraints related to computational and sequencing costs, finding the appropriate number of controls is a crucial step to identifying somatic variants. Our approach utilizes public databases for canonical somatic variants as well as germline variants and leverages information gathered about nearby positions in the normal controls. Drawing from our cohort of targeted capture panel sequencing of tumor and normal samples with varying tumortypes and demographics, these served as a benchmark for our tumor-only variant calling pipeline to observe the relationship between our ability to correctly classify variants against a number of unmatched normals. With our benchmarked samples, approximately ten normal controls were needed to maintain 94% sensitivity, 99% specificity and 76% positive predictive value, far outperforming comparable methods. Our approach, called UNMASC, also serves as a supplement to traditional tumor with matched normal variant calling workflows and can potentially extend to other concerns arising from analyzing next generation sequencing data.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


