Olusola Olalekan Elekofehinti, Opeyemi Iwaloye, Olorunfemi R Molehin, Courage D Famusiwa
{"title":"利用e -药效团模型、QSAR和分子动力学模拟鉴定针对SARS-CoV-2的3c样蛋白酶的大型天然产物文库中的先导化合物","authors":"Olusola Olalekan Elekofehinti, Opeyemi Iwaloye, Olorunfemi R Molehin, Courage D Famusiwa","doi":"10.1007/s40203-021-00109-7","DOIUrl":null,"url":null,"abstract":"<p><p>COVID-19 is a novel disease caused by SARS-CoV-2 and has made a catastrophic impact on the global economy. As it is, there is no officially FDA approved drug to alleviate the negative impact of SARS-CoV-2 on human health. Numerous drug targets for neutralizing coronavirus infection have been identified, among them is 3-chymotrypsin-like-protease (3CL<sup>pro</sup>), a viral protease responsible for the viral replication is chosen for this study. This study aimed at finding novel inhibitors of SARS-CoV-2 3C-like protease from the natural library using computational approaches. A total of 69,000 compounds from natural product library were screened to match a minimum of 3 features from the five sites e-pharmacophore model. Compounds with fitness score of 1.00 and above were consequently filtered by executing molecular docking studies via Glide docking algorithm. Qikprop also predicted the compounds drug-likeness and pharmacokinetic features; besides, the QSAR model built from KPLS analysis with radial as binary fingerprint was used to predict the compounds inhibition properties against SARS-CoV-2 3C-like protease. Fifty ns molecular dynamics (MD) simulation was carried out using GROMACS software to understand the dynamics of binding. Nine (9) lead compounds from the natural products library were discovered; seven among them were found to be more potent than lopinavir based on energies of binding. STOCK1N-98687 with docking score of -9.295 kcal/mol had considerable predicted bioactivity (4.427 µM) against SARS-CoV-2 3C-like protease and satisfactory drug-like features than the experimental drug lopinavir. Post-docking analysis by MM-GBSA confirmed the stability of STOCK1N-98687 bound 3CL<sup>pro</sup> crystal structure. MD simulation of STOCKIN-98687 with 3CL<sup>pro</sup> at 50 ns showed high stability and low fluctuation of the complex. This study revealed compound STOCK1N-98687 as potential 3CL<sup>pro</sup> inhibitor; therefore, a wet experiment is worth exploring to confirm the therapeutic potential of STOCK1N-98687 as an antiviral agent.</p>","PeriodicalId":13380,"journal":{"name":"In Silico Pharmacology","volume":" ","pages":"49"},"PeriodicalIF":0.0000,"publicationDate":"2021-08-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8349134/pdf/","citationCount":"9","resultStr":"{\"title\":\"Identification of lead compounds from large natural product library targeting 3C-like protease of SARS-CoV-2 using E-pharmacophore modelling, QSAR and molecular dynamics simulation.\",\"authors\":\"Olusola Olalekan Elekofehinti, Opeyemi Iwaloye, Olorunfemi R Molehin, Courage D Famusiwa\",\"doi\":\"10.1007/s40203-021-00109-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>COVID-19 is a novel disease caused by SARS-CoV-2 and has made a catastrophic impact on the global economy. As it is, there is no officially FDA approved drug to alleviate the negative impact of SARS-CoV-2 on human health. Numerous drug targets for neutralizing coronavirus infection have been identified, among them is 3-chymotrypsin-like-protease (3CL<sup>pro</sup>), a viral protease responsible for the viral replication is chosen for this study. This study aimed at finding novel inhibitors of SARS-CoV-2 3C-like protease from the natural library using computational approaches. A total of 69,000 compounds from natural product library were screened to match a minimum of 3 features from the five sites e-pharmacophore model. Compounds with fitness score of 1.00 and above were consequently filtered by executing molecular docking studies via Glide docking algorithm. Qikprop also predicted the compounds drug-likeness and pharmacokinetic features; besides, the QSAR model built from KPLS analysis with radial as binary fingerprint was used to predict the compounds inhibition properties against SARS-CoV-2 3C-like protease. Fifty ns molecular dynamics (MD) simulation was carried out using GROMACS software to understand the dynamics of binding. Nine (9) lead compounds from the natural products library were discovered; seven among them were found to be more potent than lopinavir based on energies of binding. STOCK1N-98687 with docking score of -9.295 kcal/mol had considerable predicted bioactivity (4.427 µM) against SARS-CoV-2 3C-like protease and satisfactory drug-like features than the experimental drug lopinavir. Post-docking analysis by MM-GBSA confirmed the stability of STOCK1N-98687 bound 3CL<sup>pro</sup> crystal structure. MD simulation of STOCKIN-98687 with 3CL<sup>pro</sup> at 50 ns showed high stability and low fluctuation of the complex. This study revealed compound STOCK1N-98687 as potential 3CL<sup>pro</sup> inhibitor; therefore, a wet experiment is worth exploring to confirm the therapeutic potential of STOCK1N-98687 as an antiviral agent.</p>\",\"PeriodicalId\":13380,\"journal\":{\"name\":\"In Silico Pharmacology\",\"volume\":\" \",\"pages\":\"49\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-08-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8349134/pdf/\",\"citationCount\":\"9\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"In Silico Pharmacology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1007/s40203-021-00109-7\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"In Silico Pharmacology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s40203-021-00109-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Identification of lead compounds from large natural product library targeting 3C-like protease of SARS-CoV-2 using E-pharmacophore modelling, QSAR and molecular dynamics simulation.
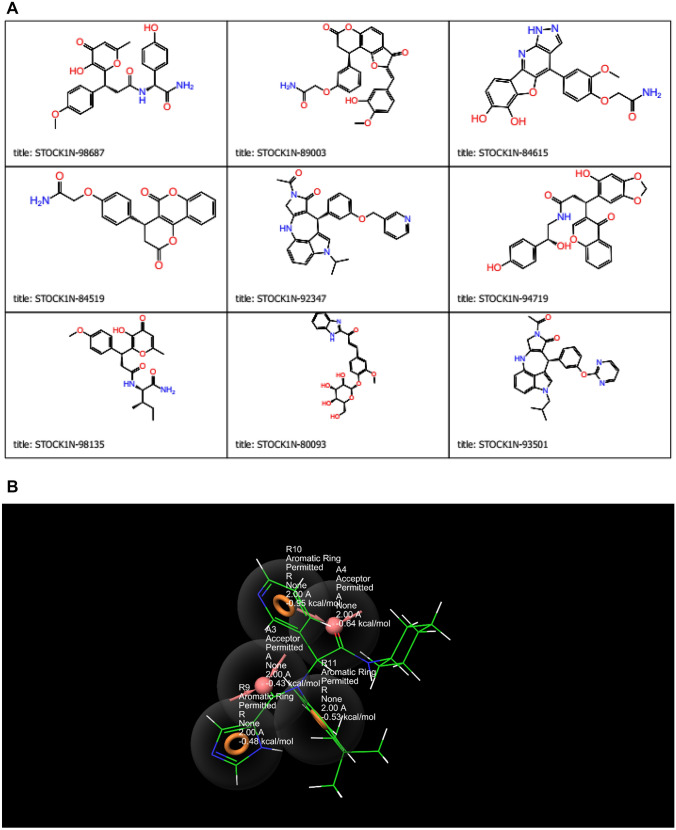
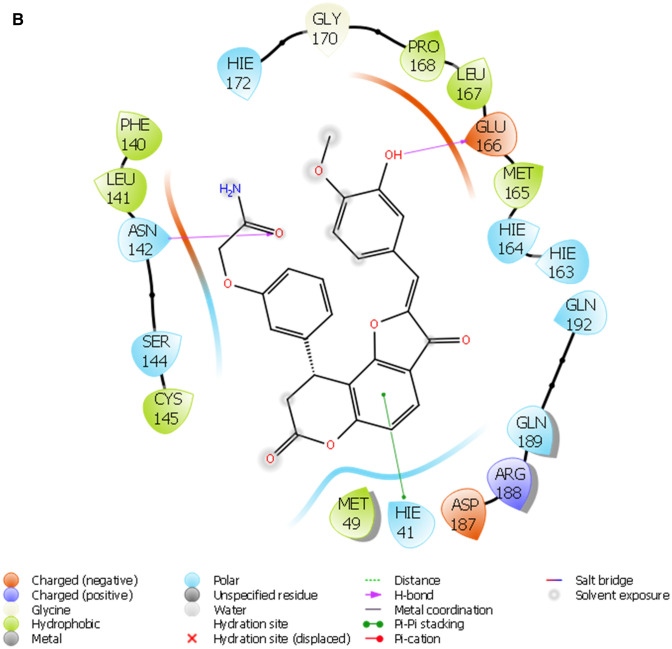
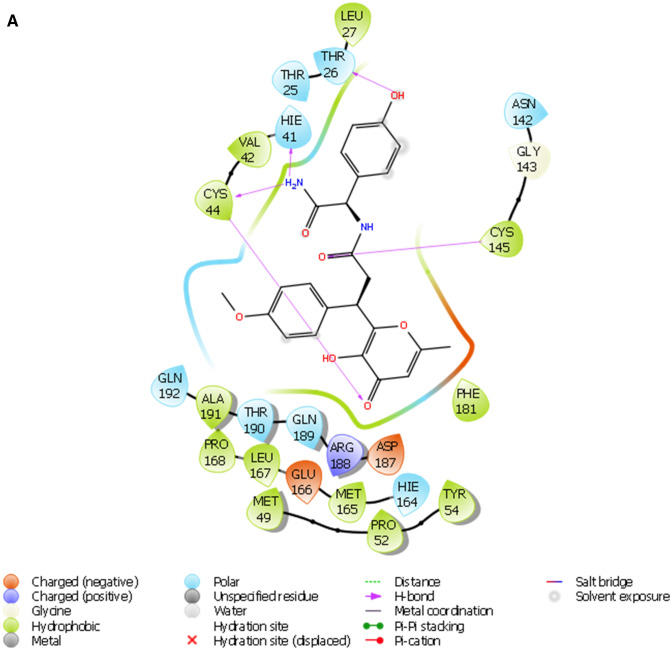
COVID-19 is a novel disease caused by SARS-CoV-2 and has made a catastrophic impact on the global economy. As it is, there is no officially FDA approved drug to alleviate the negative impact of SARS-CoV-2 on human health. Numerous drug targets for neutralizing coronavirus infection have been identified, among them is 3-chymotrypsin-like-protease (3CLpro), a viral protease responsible for the viral replication is chosen for this study. This study aimed at finding novel inhibitors of SARS-CoV-2 3C-like protease from the natural library using computational approaches. A total of 69,000 compounds from natural product library were screened to match a minimum of 3 features from the five sites e-pharmacophore model. Compounds with fitness score of 1.00 and above were consequently filtered by executing molecular docking studies via Glide docking algorithm. Qikprop also predicted the compounds drug-likeness and pharmacokinetic features; besides, the QSAR model built from KPLS analysis with radial as binary fingerprint was used to predict the compounds inhibition properties against SARS-CoV-2 3C-like protease. Fifty ns molecular dynamics (MD) simulation was carried out using GROMACS software to understand the dynamics of binding. Nine (9) lead compounds from the natural products library were discovered; seven among them were found to be more potent than lopinavir based on energies of binding. STOCK1N-98687 with docking score of -9.295 kcal/mol had considerable predicted bioactivity (4.427 µM) against SARS-CoV-2 3C-like protease and satisfactory drug-like features than the experimental drug lopinavir. Post-docking analysis by MM-GBSA confirmed the stability of STOCK1N-98687 bound 3CLpro crystal structure. MD simulation of STOCKIN-98687 with 3CLpro at 50 ns showed high stability and low fluctuation of the complex. This study revealed compound STOCK1N-98687 as potential 3CLpro inhibitor; therefore, a wet experiment is worth exploring to confirm the therapeutic potential of STOCK1N-98687 as an antiviral agent.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


