David Schaller, Manuela Geiß, Marc Hellmuth, Peter F Stadler
{"title":"最佳匹配图编辑的启发式算法。","authors":"David Schaller, Manuela Geiß, Marc Hellmuth, Peter F Stadler","doi":"10.1186/s13015-021-00196-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Best match graphs (BMGs) are a class of colored digraphs that naturally appear in mathematical phylogenetics as a representation of the pairwise most closely related genes among multiple species. An arc connects a gene x with a gene y from another species (vertex color) Y whenever it is one of the phylogenetically closest relatives of x. BMGs can be approximated with the help of similarity measures between gene sequences, albeit not without errors. Empirical estimates thus will usually violate the theoretical properties of BMGs. The corresponding graph editing problem can be used to guide error correction for best match data. Since the arc set modification problems for BMGs are NP-complete, efficient heuristics are needed if BMGs are to be used for the practical analysis of biological sequence data.</p><p><strong>Results: </strong>Since BMGs have a characterization in terms of consistency of a certain set of rooted triples (binary trees on three vertices) defined on the set of genes, we consider heuristics that operate on triple sets. As an alternative, we show that there is a close connection to a set partitioning problem that leads to a class of top-down recursive algorithms that are similar to Aho's supertree algorithm and give rise to BMG editing algorithms that are consistent in the sense that they leave BMGs invariant. Extensive benchmarking shows that community detection algorithms for the partitioning steps perform best for BMG editing.</p><p><strong>Conclusion: </strong>Noisy BMG data can be corrected with sufficient accuracy and efficiency to make BMGs an attractive alternative to classical phylogenetic methods.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"16 1","pages":"19"},"PeriodicalIF":1.7000,"publicationDate":"2021-08-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8369769/pdf/","citationCount":"5","resultStr":"{\"title\":\"Heuristic algorithms for best match graph editing.\",\"authors\":\"David Schaller, Manuela Geiß, Marc Hellmuth, Peter F Stadler\",\"doi\":\"10.1186/s13015-021-00196-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Best match graphs (BMGs) are a class of colored digraphs that naturally appear in mathematical phylogenetics as a representation of the pairwise most closely related genes among multiple species. An arc connects a gene x with a gene y from another species (vertex color) Y whenever it is one of the phylogenetically closest relatives of x. BMGs can be approximated with the help of similarity measures between gene sequences, albeit not without errors. Empirical estimates thus will usually violate the theoretical properties of BMGs. The corresponding graph editing problem can be used to guide error correction for best match data. Since the arc set modification problems for BMGs are NP-complete, efficient heuristics are needed if BMGs are to be used for the practical analysis of biological sequence data.</p><p><strong>Results: </strong>Since BMGs have a characterization in terms of consistency of a certain set of rooted triples (binary trees on three vertices) defined on the set of genes, we consider heuristics that operate on triple sets. As an alternative, we show that there is a close connection to a set partitioning problem that leads to a class of top-down recursive algorithms that are similar to Aho's supertree algorithm and give rise to BMG editing algorithms that are consistent in the sense that they leave BMGs invariant. Extensive benchmarking shows that community detection algorithms for the partitioning steps perform best for BMG editing.</p><p><strong>Conclusion: </strong>Noisy BMG data can be corrected with sufficient accuracy and efficiency to make BMGs an attractive alternative to classical phylogenetic methods.</p>\",\"PeriodicalId\":50823,\"journal\":{\"name\":\"Algorithms for Molecular Biology\",\"volume\":\"16 1\",\"pages\":\"19\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2021-08-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8369769/pdf/\",\"citationCount\":\"5\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Algorithms for Molecular Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13015-021-00196-3\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-021-00196-3","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Heuristic algorithms for best match graph editing.
Background: Best match graphs (BMGs) are a class of colored digraphs that naturally appear in mathematical phylogenetics as a representation of the pairwise most closely related genes among multiple species. An arc connects a gene x with a gene y from another species (vertex color) Y whenever it is one of the phylogenetically closest relatives of x. BMGs can be approximated with the help of similarity measures between gene sequences, albeit not without errors. Empirical estimates thus will usually violate the theoretical properties of BMGs. The corresponding graph editing problem can be used to guide error correction for best match data. Since the arc set modification problems for BMGs are NP-complete, efficient heuristics are needed if BMGs are to be used for the practical analysis of biological sequence data.
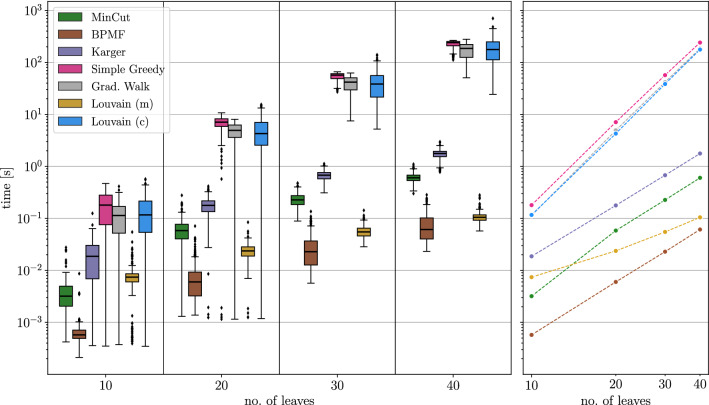
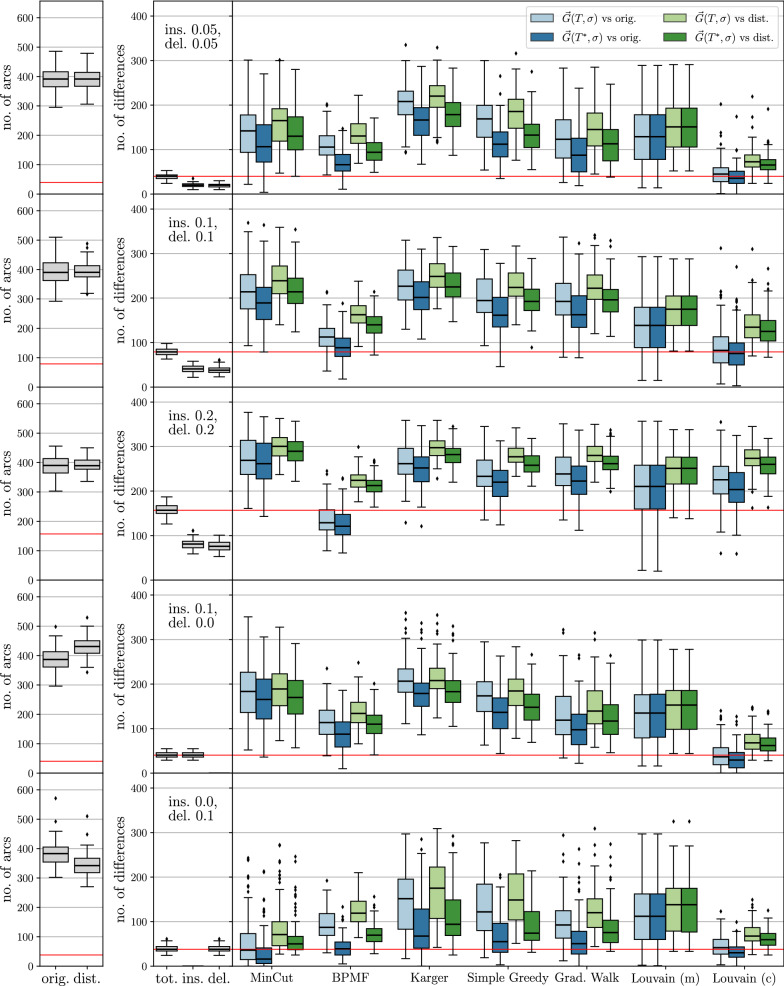
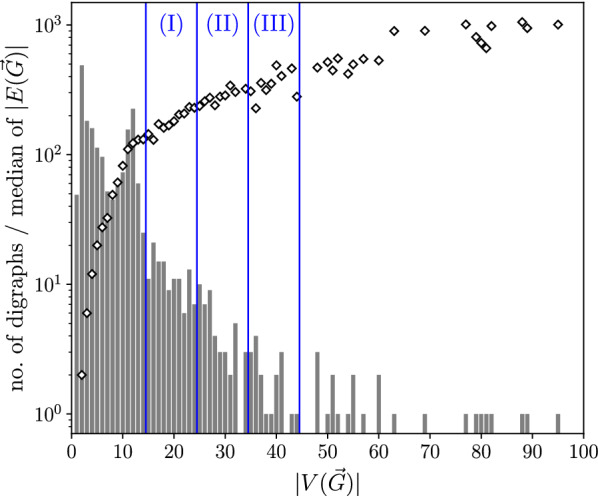
Results: Since BMGs have a characterization in terms of consistency of a certain set of rooted triples (binary trees on three vertices) defined on the set of genes, we consider heuristics that operate on triple sets. As an alternative, we show that there is a close connection to a set partitioning problem that leads to a class of top-down recursive algorithms that are similar to Aho's supertree algorithm and give rise to BMG editing algorithms that are consistent in the sense that they leave BMGs invariant. Extensive benchmarking shows that community detection algorithms for the partitioning steps perform best for BMG editing.
Conclusion: Noisy BMG data can be corrected with sufficient accuracy and efficiency to make BMGs an attractive alternative to classical phylogenetic methods.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


