Daniel Zhao, Daniel Y Kim, Peter Chen, Patrick Yu, Sophia Ho, Stephanie W Cheng, Cindy Zhao, Jimmy A Guo, Yun R Li
{"title":"泛肿瘤生存分类与临床病理和靶向基因表达特征。","authors":"Daniel Zhao, Daniel Y Kim, Peter Chen, Patrick Yu, Sophia Ho, Stephanie W Cheng, Cindy Zhao, Jimmy A Guo, Yun R Li","doi":"10.1177/11769351211035137","DOIUrl":null,"url":null,"abstract":"<p><p>Prognostication for patients with cancer is important for clinical planning and management, but remains challenging given the large number of factors that can influence outcomes. As such, there is a need to identify features that can robustly predict patient outcomes. We evaluated 8608 patient tumor samples across 16 cancer types from The Cancer Genome Atlas and generated distinct survival classifiers for each using clinical and histopathological data accessible to standard oncology workflows. For cancers that had poor model performance, we deployed a random-forest-embedded sequential forward selection approach that began with an initial subset of the 15 most predictive clinicopathological features before sequentially appending the next most informative gene as an additional feature. With classifiers derived from clinical and histopathological features alone, we observed cancer-type-dependent model performance and an area under the receiver operating curve (AUROC) range of 0.65 to 0.91 across all 16 cancer types for 1- and 3-year survival prediction, with some classifiers consistently outperforming those for others. As such, for cancers that had poor model performance, we posited that the addition of more complex biomolecular features could enhance our ability to prognose patients where clinicopathological features were insufficient. With the inclusion of gene expression data, model performance for 3 select cancers (glioblastoma, stomach/gastric adenocarcinoma, ovarian serous carcinoma) markedly increased from initial AUROC scores of 0.66, 0.69, and 0.67 to 0.76, 0.77, and 0.77, respectively. As a whole, this study provides a thorough examination of the relative contributions of clinical, pathological, and gene expression data in predicting overall survival and reveals cancer types for which clinical features are already strong predictors and those where additional biomolecular information is needed.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":"20 ","pages":"11769351211035137"},"PeriodicalIF":2.4000,"publicationDate":"2021-07-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1177/11769351211035137","citationCount":"3","resultStr":"{\"title\":\"Pan-Cancer Survival Classification With Clinicopathological and Targeted Gene Expression Features.\",\"authors\":\"Daniel Zhao, Daniel Y Kim, Peter Chen, Patrick Yu, Sophia Ho, Stephanie W Cheng, Cindy Zhao, Jimmy A Guo, Yun R Li\",\"doi\":\"10.1177/11769351211035137\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Prognostication for patients with cancer is important for clinical planning and management, but remains challenging given the large number of factors that can influence outcomes. As such, there is a need to identify features that can robustly predict patient outcomes. We evaluated 8608 patient tumor samples across 16 cancer types from The Cancer Genome Atlas and generated distinct survival classifiers for each using clinical and histopathological data accessible to standard oncology workflows. For cancers that had poor model performance, we deployed a random-forest-embedded sequential forward selection approach that began with an initial subset of the 15 most predictive clinicopathological features before sequentially appending the next most informative gene as an additional feature. With classifiers derived from clinical and histopathological features alone, we observed cancer-type-dependent model performance and an area under the receiver operating curve (AUROC) range of 0.65 to 0.91 across all 16 cancer types for 1- and 3-year survival prediction, with some classifiers consistently outperforming those for others. As such, for cancers that had poor model performance, we posited that the addition of more complex biomolecular features could enhance our ability to prognose patients where clinicopathological features were insufficient. With the inclusion of gene expression data, model performance for 3 select cancers (glioblastoma, stomach/gastric adenocarcinoma, ovarian serous carcinoma) markedly increased from initial AUROC scores of 0.66, 0.69, and 0.67 to 0.76, 0.77, and 0.77, respectively. As a whole, this study provides a thorough examination of the relative contributions of clinical, pathological, and gene expression data in predicting overall survival and reveals cancer types for which clinical features are already strong predictors and those where additional biomolecular information is needed.</p>\",\"PeriodicalId\":35418,\"journal\":{\"name\":\"Cancer Informatics\",\"volume\":\"20 \",\"pages\":\"11769351211035137\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2021-07-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1177/11769351211035137\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cancer Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11769351211035137\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351211035137","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Pan-Cancer Survival Classification With Clinicopathological and Targeted Gene Expression Features.
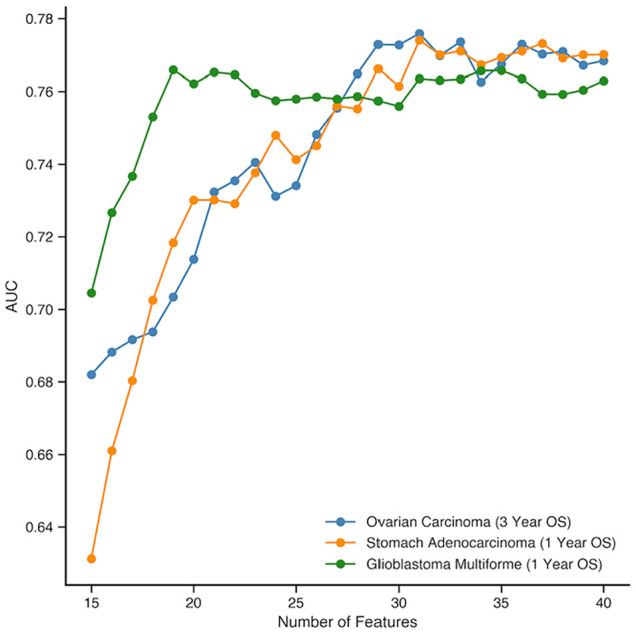
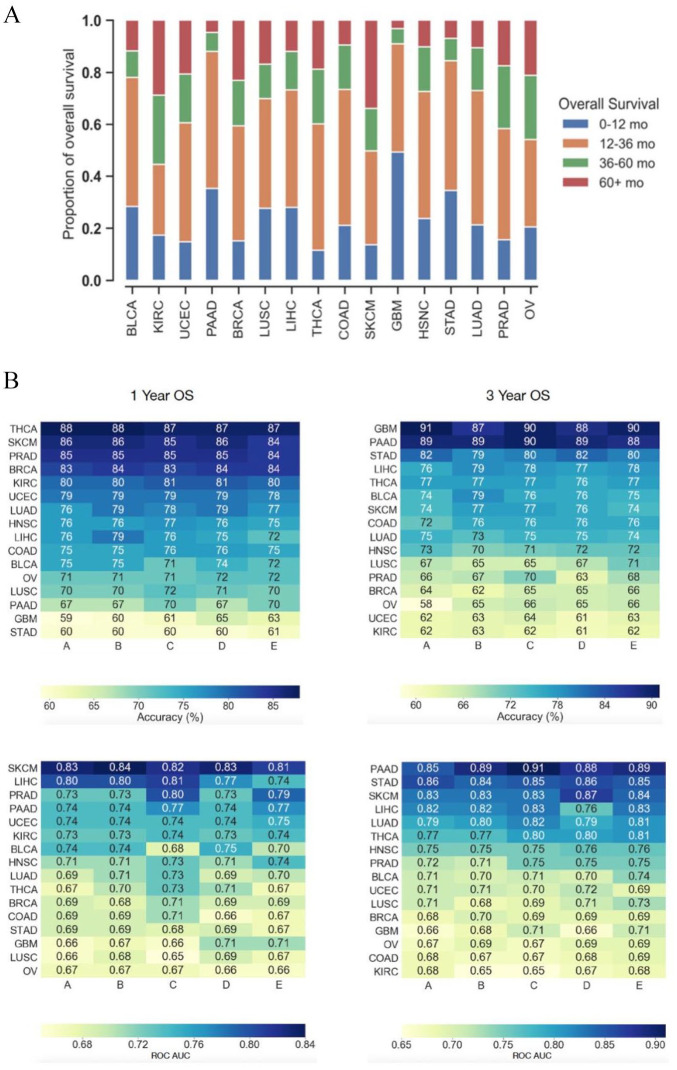
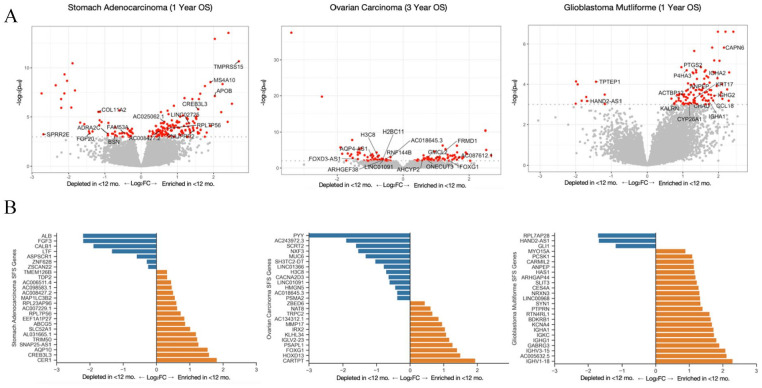
Prognostication for patients with cancer is important for clinical planning and management, but remains challenging given the large number of factors that can influence outcomes. As such, there is a need to identify features that can robustly predict patient outcomes. We evaluated 8608 patient tumor samples across 16 cancer types from The Cancer Genome Atlas and generated distinct survival classifiers for each using clinical and histopathological data accessible to standard oncology workflows. For cancers that had poor model performance, we deployed a random-forest-embedded sequential forward selection approach that began with an initial subset of the 15 most predictive clinicopathological features before sequentially appending the next most informative gene as an additional feature. With classifiers derived from clinical and histopathological features alone, we observed cancer-type-dependent model performance and an area under the receiver operating curve (AUROC) range of 0.65 to 0.91 across all 16 cancer types for 1- and 3-year survival prediction, with some classifiers consistently outperforming those for others. As such, for cancers that had poor model performance, we posited that the addition of more complex biomolecular features could enhance our ability to prognose patients where clinicopathological features were insufficient. With the inclusion of gene expression data, model performance for 3 select cancers (glioblastoma, stomach/gastric adenocarcinoma, ovarian serous carcinoma) markedly increased from initial AUROC scores of 0.66, 0.69, and 0.67 to 0.76, 0.77, and 0.77, respectively. As a whole, this study provides a thorough examination of the relative contributions of clinical, pathological, and gene expression data in predicting overall survival and reveals cancer types for which clinical features are already strong predictors and those where additional biomolecular information is needed.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


