Lorenzo R Sewanan, Jinkyu Park, Michael J Rynkiewicz, Alice W Racca, Nikolaos Papoutsidakis, Jonas Schwan, Daniel L Jacoby, Jeffrey R Moore, William Lehman, Yibing Qyang, Stuart G Campbell
{"title":"在携带TPM1 E192K突变的患者中,丧失过桥抑制可导致病理性心肌肥大。","authors":"Lorenzo R Sewanan, Jinkyu Park, Michael J Rynkiewicz, Alice W Racca, Nikolaos Papoutsidakis, Jonas Schwan, Daniel L Jacoby, Jeffrey R Moore, William Lehman, Yibing Qyang, Stuart G Campbell","doi":"10.1085/jgp.202012640","DOIUrl":null,"url":null,"abstract":"<p><p>Hypertrophic cardiomyopathy (HCM) is an inherited disorder caused primarily by mutations to thick and thinfilament proteins. Although thin filament mutations are less prevalent than their oft-studied thick filament counterparts, they are frequently associated with severe patient phenotypes and can offer important insight into fundamental disease mechanisms. We have performed a detailed study of tropomyosin (TPM1) E192K, a variant of uncertain significance associated with HCM. Molecular dynamics revealed that E192K results in a more flexible TPM1 molecule, which could affect its ability to regulate crossbridges. In vitro motility assays of regulated actin filaments containing TPM1 E192K showed an overall loss of Ca2+ sensitivity. To understand these effects, we used multiscale computational models that suggested a subtle phenotype in which E192K leads to an inability to completely inhibit actin-myosin crossbridge activity at low Ca2+. To assess the physiological impact of the mutation, we generated patient-derived engineered heart tissues expressing E192K. These tissues showed disease features similar to those of the patients, including cellular hypertrophy, hypercontractility, and diastolic dysfunction. We hypothesized that excess residual crossbridge activity could be triggering cellular hypertrophy, even if the overall Ca2+ sensitivity was reduced by E192K. To test this hypothesis, the cardiac myosin-specific inhibitor mavacamten was applied to patient-derived engineered heart tissues for 4 d followed by 24 h of washout. Chronic mavacamten treatment abolished contractile differences between control and TPM1 E192K engineered heart tissues and reversed hypertrophy in cardiomyocytes. These results suggest that the TPM1 E192K mutation triggers cardiomyocyte hypertrophy by permitting excess residual crossbridge activity. These studies also provide direct evidence that myosin inhibition by mavacamten can counteract the hypertrophic effects of mutant tropomyosin.</p>","PeriodicalId":173753,"journal":{"name":"The Journal of General Physiology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2021-09-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/10/66/JGP_202012640.PMC8321830.pdf","citationCount":"13","resultStr":"{\"title\":\"Loss of crossbridge inhibition drives pathological cardiac hypertrophy in patients harboring the TPM1 E192K mutation.\",\"authors\":\"Lorenzo R Sewanan, Jinkyu Park, Michael J Rynkiewicz, Alice W Racca, Nikolaos Papoutsidakis, Jonas Schwan, Daniel L Jacoby, Jeffrey R Moore, William Lehman, Yibing Qyang, Stuart G Campbell\",\"doi\":\"10.1085/jgp.202012640\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hypertrophic cardiomyopathy (HCM) is an inherited disorder caused primarily by mutations to thick and thinfilament proteins. Although thin filament mutations are less prevalent than their oft-studied thick filament counterparts, they are frequently associated with severe patient phenotypes and can offer important insight into fundamental disease mechanisms. We have performed a detailed study of tropomyosin (TPM1) E192K, a variant of uncertain significance associated with HCM. Molecular dynamics revealed that E192K results in a more flexible TPM1 molecule, which could affect its ability to regulate crossbridges. In vitro motility assays of regulated actin filaments containing TPM1 E192K showed an overall loss of Ca2+ sensitivity. To understand these effects, we used multiscale computational models that suggested a subtle phenotype in which E192K leads to an inability to completely inhibit actin-myosin crossbridge activity at low Ca2+. To assess the physiological impact of the mutation, we generated patient-derived engineered heart tissues expressing E192K. These tissues showed disease features similar to those of the patients, including cellular hypertrophy, hypercontractility, and diastolic dysfunction. We hypothesized that excess residual crossbridge activity could be triggering cellular hypertrophy, even if the overall Ca2+ sensitivity was reduced by E192K. To test this hypothesis, the cardiac myosin-specific inhibitor mavacamten was applied to patient-derived engineered heart tissues for 4 d followed by 24 h of washout. Chronic mavacamten treatment abolished contractile differences between control and TPM1 E192K engineered heart tissues and reversed hypertrophy in cardiomyocytes. These results suggest that the TPM1 E192K mutation triggers cardiomyocyte hypertrophy by permitting excess residual crossbridge activity. These studies also provide direct evidence that myosin inhibition by mavacamten can counteract the hypertrophic effects of mutant tropomyosin.</p>\",\"PeriodicalId\":173753,\"journal\":{\"name\":\"The Journal of General Physiology\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-09-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/10/66/JGP_202012640.PMC8321830.pdf\",\"citationCount\":\"13\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of General Physiology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1085/jgp.202012640\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/7/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of General Physiology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1085/jgp.202012640","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/7/28 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Loss of crossbridge inhibition drives pathological cardiac hypertrophy in patients harboring the TPM1 E192K mutation.
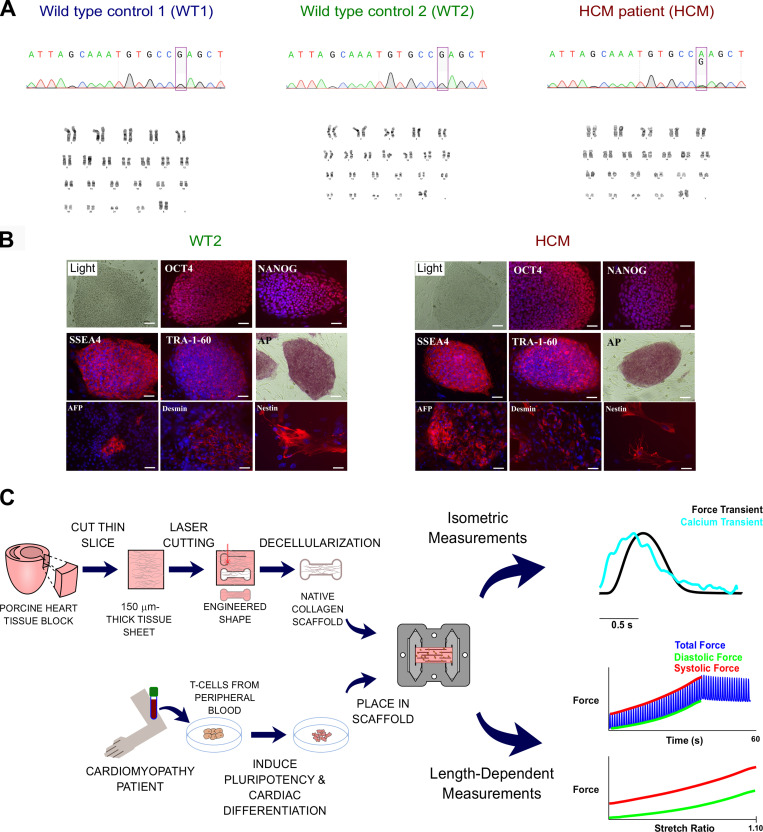
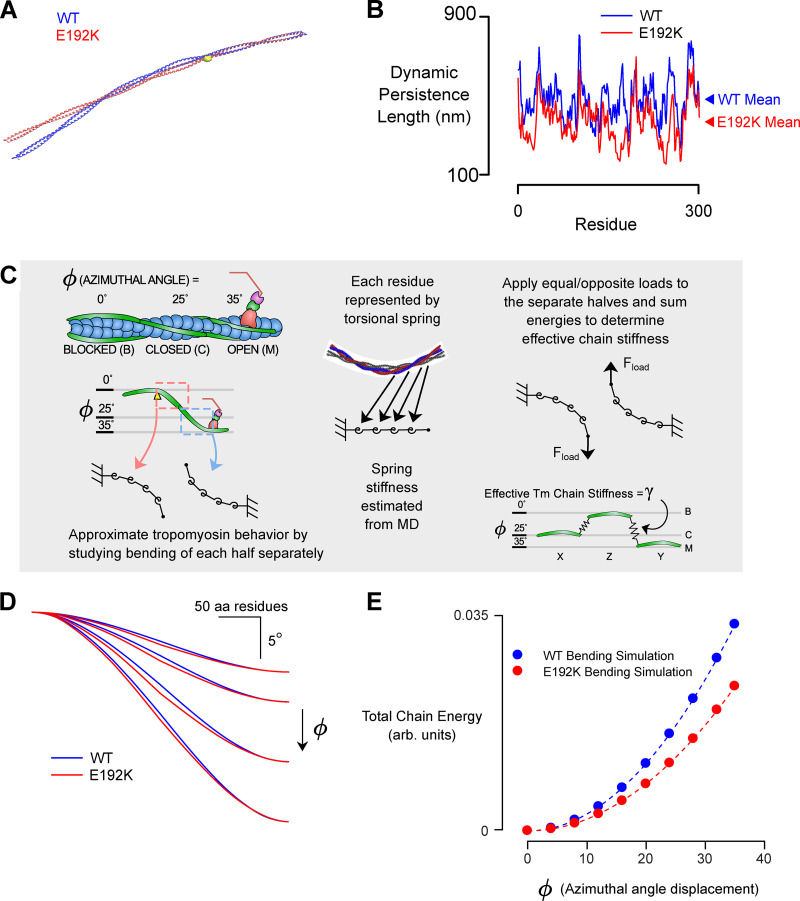
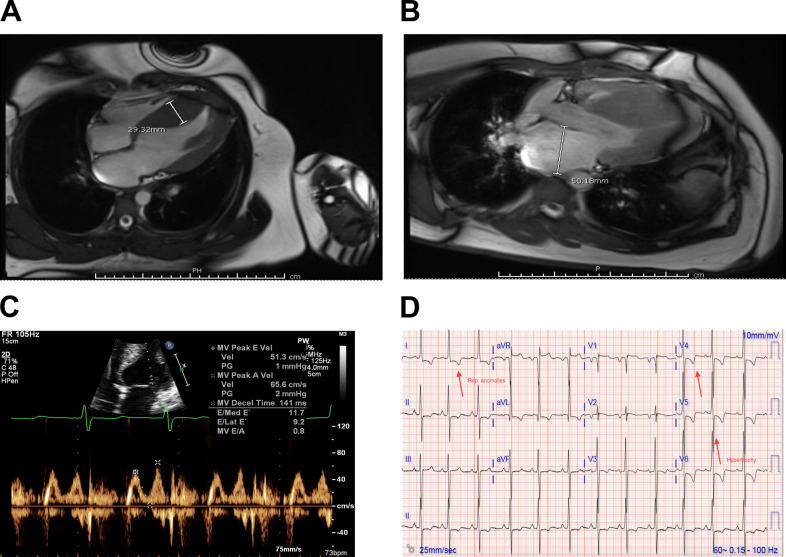
Hypertrophic cardiomyopathy (HCM) is an inherited disorder caused primarily by mutations to thick and thinfilament proteins. Although thin filament mutations are less prevalent than their oft-studied thick filament counterparts, they are frequently associated with severe patient phenotypes and can offer important insight into fundamental disease mechanisms. We have performed a detailed study of tropomyosin (TPM1) E192K, a variant of uncertain significance associated with HCM. Molecular dynamics revealed that E192K results in a more flexible TPM1 molecule, which could affect its ability to regulate crossbridges. In vitro motility assays of regulated actin filaments containing TPM1 E192K showed an overall loss of Ca2+ sensitivity. To understand these effects, we used multiscale computational models that suggested a subtle phenotype in which E192K leads to an inability to completely inhibit actin-myosin crossbridge activity at low Ca2+. To assess the physiological impact of the mutation, we generated patient-derived engineered heart tissues expressing E192K. These tissues showed disease features similar to those of the patients, including cellular hypertrophy, hypercontractility, and diastolic dysfunction. We hypothesized that excess residual crossbridge activity could be triggering cellular hypertrophy, even if the overall Ca2+ sensitivity was reduced by E192K. To test this hypothesis, the cardiac myosin-specific inhibitor mavacamten was applied to patient-derived engineered heart tissues for 4 d followed by 24 h of washout. Chronic mavacamten treatment abolished contractile differences between control and TPM1 E192K engineered heart tissues and reversed hypertrophy in cardiomyocytes. These results suggest that the TPM1 E192K mutation triggers cardiomyocyte hypertrophy by permitting excess residual crossbridge activity. These studies also provide direct evidence that myosin inhibition by mavacamten can counteract the hypertrophic effects of mutant tropomyosin.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


