Jacob P Miller, Hans J Moldenhauer, Sotirios Keros, Andrea L Meredith
{"title":"新出现的 KCNMA1 相关通道病变异谱和临床特征。","authors":"Jacob P Miller, Hans J Moldenhauer, Sotirios Keros, Andrea L Meredith","doi":"10.1080/19336950.2021.1938852","DOIUrl":null,"url":null,"abstract":"<p><p><i>KCNMA1</i>-linked channelopathy is an emerging neurological disorder characterized by heterogeneous and overlapping combinations of movement disorder, seizure, developmental delay, and intellectual disability. <i>KCNMA1</i> encodes the BK K<sup>+</sup> channel, which contributes to both excitatory and inhibitory neuronal and muscle activity. Understanding the basis of the disorder is an important area of active investigation; however, the rare prevalence has hampered the development of large patient cohorts necessary to establish genotype-phenotype correlations. In this review, we summarize 37 <i>KCNMA1</i> alleles from 69 patients currently defining the channelopathy and assess key diagnostic and clinical hallmarks. At present, 3 variants are classified as gain-of-function with respect to BK channel activity, 14 loss-of-function, 15 variants of uncertain significance, and putative benign/VUS. Symptoms associated with these variants were curated from patient-provided information and prior publications to define the spectrum of clinical phenotypes. In this newly expanded cohort, seizures showed no differential distribution between patients harboring GOF and LOF variants, while movement disorders segregated by mutation type. Paroxysmal non-kinesigenic dyskinesia was predominantly observed among patients with GOF alleles of the BK channel, although not exclusively so, while additional movement disorders were observed in patients with LOF variants. Neurodevelopmental and structural brain abnormalities were prevalent in patients with LOF mutations. In contrast to mutations, disease-associated <i>KCNMA1</i> single nucleotide polymorphisms were not predominantly related to neurological phenotypes but covered a wider set of peripheral physiological functions. Together, this review provides additional evidence exploring the genetic and biochemical basis for <i>KCNMA1-</i>linked channelopathy and summarizes the clinical repository of patient symptoms across multiple types of <i>KCNMA1</i> gene variants.</p>","PeriodicalId":72555,"journal":{"name":"Channels (Austin, Tex.)","volume":" ","pages":"447-464"},"PeriodicalIF":0.0000,"publicationDate":"2021-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8259716/pdf/","citationCount":"0","resultStr":"{\"title\":\"An emerging spectrum of variants and clinical features in <i>KCNMA1</i>-linked channelopathy.\",\"authors\":\"Jacob P Miller, Hans J Moldenhauer, Sotirios Keros, Andrea L Meredith\",\"doi\":\"10.1080/19336950.2021.1938852\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p><i>KCNMA1</i>-linked channelopathy is an emerging neurological disorder characterized by heterogeneous and overlapping combinations of movement disorder, seizure, developmental delay, and intellectual disability. <i>KCNMA1</i> encodes the BK K<sup>+</sup> channel, which contributes to both excitatory and inhibitory neuronal and muscle activity. Understanding the basis of the disorder is an important area of active investigation; however, the rare prevalence has hampered the development of large patient cohorts necessary to establish genotype-phenotype correlations. In this review, we summarize 37 <i>KCNMA1</i> alleles from 69 patients currently defining the channelopathy and assess key diagnostic and clinical hallmarks. At present, 3 variants are classified as gain-of-function with respect to BK channel activity, 14 loss-of-function, 15 variants of uncertain significance, and putative benign/VUS. Symptoms associated with these variants were curated from patient-provided information and prior publications to define the spectrum of clinical phenotypes. In this newly expanded cohort, seizures showed no differential distribution between patients harboring GOF and LOF variants, while movement disorders segregated by mutation type. Paroxysmal non-kinesigenic dyskinesia was predominantly observed among patients with GOF alleles of the BK channel, although not exclusively so, while additional movement disorders were observed in patients with LOF variants. Neurodevelopmental and structural brain abnormalities were prevalent in patients with LOF mutations. In contrast to mutations, disease-associated <i>KCNMA1</i> single nucleotide polymorphisms were not predominantly related to neurological phenotypes but covered a wider set of peripheral physiological functions. Together, this review provides additional evidence exploring the genetic and biochemical basis for <i>KCNMA1-</i>linked channelopathy and summarizes the clinical repository of patient symptoms across multiple types of <i>KCNMA1</i> gene variants.</p>\",\"PeriodicalId\":72555,\"journal\":{\"name\":\"Channels (Austin, Tex.)\",\"volume\":\" \",\"pages\":\"447-464\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8259716/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Channels (Austin, Tex.)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1080/19336950.2021.1938852\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Channels (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/19336950.2021.1938852","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
An emerging spectrum of variants and clinical features in KCNMA1-linked channelopathy.
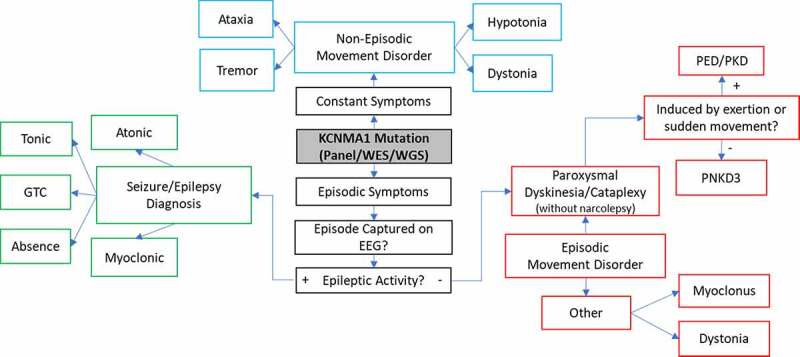
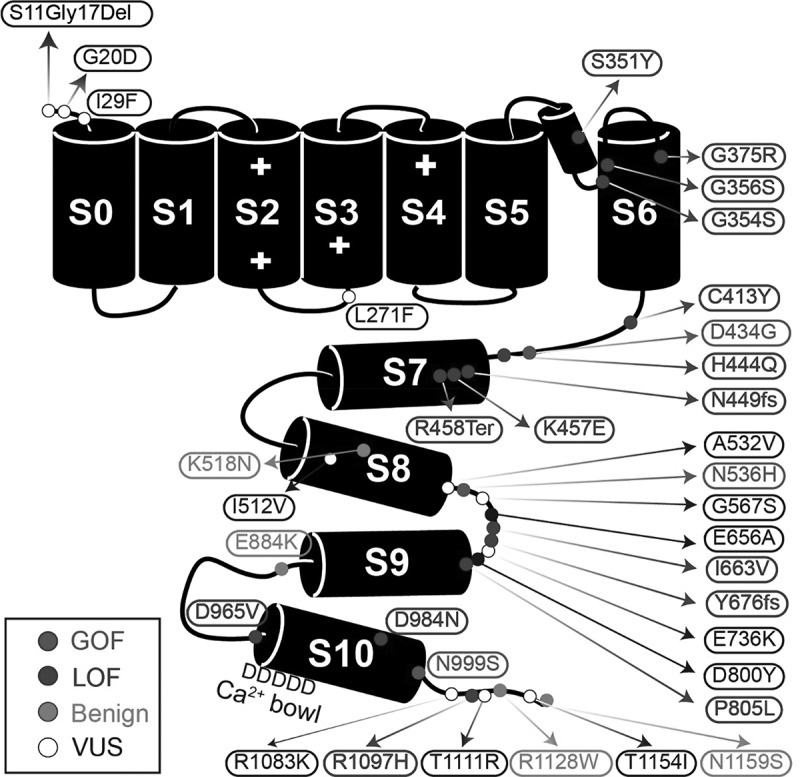
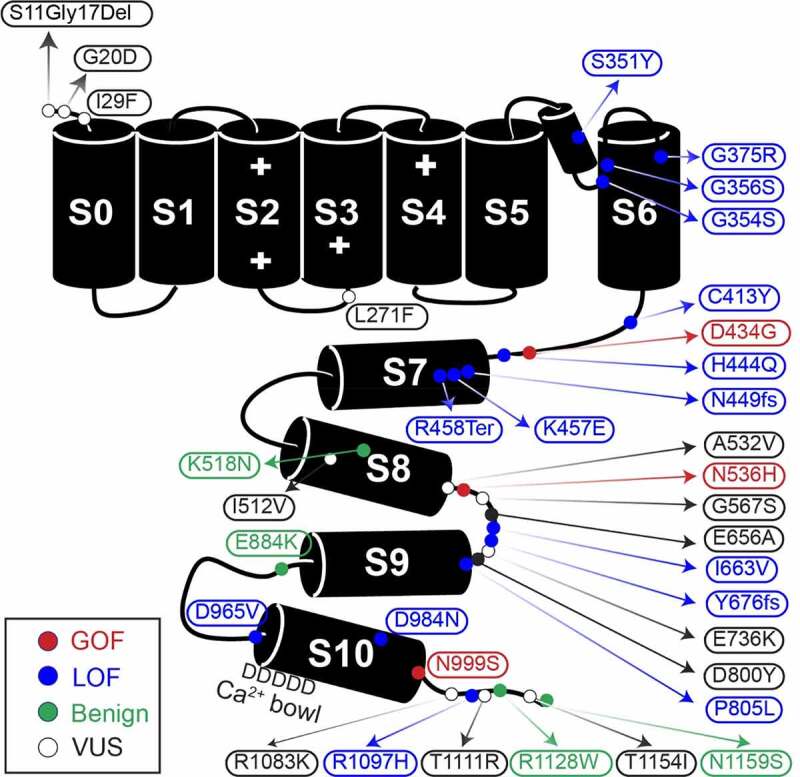
KCNMA1-linked channelopathy is an emerging neurological disorder characterized by heterogeneous and overlapping combinations of movement disorder, seizure, developmental delay, and intellectual disability. KCNMA1 encodes the BK K+ channel, which contributes to both excitatory and inhibitory neuronal and muscle activity. Understanding the basis of the disorder is an important area of active investigation; however, the rare prevalence has hampered the development of large patient cohorts necessary to establish genotype-phenotype correlations. In this review, we summarize 37 KCNMA1 alleles from 69 patients currently defining the channelopathy and assess key diagnostic and clinical hallmarks. At present, 3 variants are classified as gain-of-function with respect to BK channel activity, 14 loss-of-function, 15 variants of uncertain significance, and putative benign/VUS. Symptoms associated with these variants were curated from patient-provided information and prior publications to define the spectrum of clinical phenotypes. In this newly expanded cohort, seizures showed no differential distribution between patients harboring GOF and LOF variants, while movement disorders segregated by mutation type. Paroxysmal non-kinesigenic dyskinesia was predominantly observed among patients with GOF alleles of the BK channel, although not exclusively so, while additional movement disorders were observed in patients with LOF variants. Neurodevelopmental and structural brain abnormalities were prevalent in patients with LOF mutations. In contrast to mutations, disease-associated KCNMA1 single nucleotide polymorphisms were not predominantly related to neurological phenotypes but covered a wider set of peripheral physiological functions. Together, this review provides additional evidence exploring the genetic and biochemical basis for KCNMA1-linked channelopathy and summarizes the clinical repository of patient symptoms across multiple types of KCNMA1 gene variants.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


