Quanquan Wang, Zhe Zhao, Hongrui Shen, Qi Bing, Nan Li, Jing Hu
{"title":"五个原发性周期性麻痹家族的临床和遗传异质性分析。","authors":"Quanquan Wang, Zhe Zhao, Hongrui Shen, Qi Bing, Nan Li, Jing Hu","doi":"10.1080/19336950.2020.1857980","DOIUrl":null,"url":null,"abstract":"<p><p>To explore the clinical and genetic characteristics of five families with primary periodic paralysis (PPP). We reviewed clinical manifestations, laboratory results, electrocardiogram, electromyography, muscle biopsy, and genetic analysis from five families with PPP. Five families with PPP included: hypokalemic periodic paralysis type 1 (HypoPP1, <i>CACNA1S</i>, 1/5), hypokalemic periodic paralysis type 2 (HypoPP2, <i>SCN4A</i>, 2/5), normokalemic periodic paralysis (NormoPP, <i>SCN4A</i>, 1/5), and Andersen-Tawil syndrome (ATS, <i>KCNJ2</i>, 1/5). The basic clinical manifestations of five families were consistent with PPP, presenting with paroxysmal muscle weakness, with or without abnormal serum potassium. ATS was accompanied by ventricular arrhythmias, and skeletal and craniofacial anomalies, developing with a permanent fixed myopathy later. The electromyography showed diffuse myopathic discharge, and muscle biopsy showed tubular aggregates. Genetic testing revealed five families with PPP carried <i>CACNA1S</i> (R1242S), <i>SCN4A</i> (R675Q, T704M), and <i>KCNJ2</i> (R218Q) respectively. The novel heterozygous R1242S mutation in <i>CACNA1S</i> caused a conformational change in the protein structure, and the amino acid of this mutation site was highly conserved among different species. <i>SCN4A</i> mutations led to two phenotypes of HypoPP2 and NormoPP. PPPs are autosomal dominant disorders of ion channel dysfunction characterized by episodic flaccid muscle weakness secondary to abnormal sarcolemmal excitability. PPPs are caused by mutations in skeletal muscle calcium channel Ca<sub>V</sub>1.1 gene (<i>CACNA1S</i>), sodium channel Na<sub>V</sub>1.4 gene (<i>SCN4A</i>), and potassium channels Kir2.1, Kir3.4 genes (<i>KCNJ2, KCNJ5</i>), including HypoPP1, HypoPP2, NormoPP, HyperPP, and ATS, which have significant clinical and genetic heterogeneity. Diagnosis is based on the characteristic clinical presentation then confirmed by genetic testing.</p>","PeriodicalId":72555,"journal":{"name":"Channels (Austin, Tex.)","volume":" ","pages":"20-30"},"PeriodicalIF":0.0000,"publicationDate":"2021-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7757828/pdf/","citationCount":"0","resultStr":"{\"title\":\"The clinical and genetic heterogeneity analysis of five families with primary periodic paralysis.\",\"authors\":\"Quanquan Wang, Zhe Zhao, Hongrui Shen, Qi Bing, Nan Li, Jing Hu\",\"doi\":\"10.1080/19336950.2020.1857980\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>To explore the clinical and genetic characteristics of five families with primary periodic paralysis (PPP). We reviewed clinical manifestations, laboratory results, electrocardiogram, electromyography, muscle biopsy, and genetic analysis from five families with PPP. Five families with PPP included: hypokalemic periodic paralysis type 1 (HypoPP1, <i>CACNA1S</i>, 1/5), hypokalemic periodic paralysis type 2 (HypoPP2, <i>SCN4A</i>, 2/5), normokalemic periodic paralysis (NormoPP, <i>SCN4A</i>, 1/5), and Andersen-Tawil syndrome (ATS, <i>KCNJ2</i>, 1/5). The basic clinical manifestations of five families were consistent with PPP, presenting with paroxysmal muscle weakness, with or without abnormal serum potassium. ATS was accompanied by ventricular arrhythmias, and skeletal and craniofacial anomalies, developing with a permanent fixed myopathy later. The electromyography showed diffuse myopathic discharge, and muscle biopsy showed tubular aggregates. Genetic testing revealed five families with PPP carried <i>CACNA1S</i> (R1242S), <i>SCN4A</i> (R675Q, T704M), and <i>KCNJ2</i> (R218Q) respectively. The novel heterozygous R1242S mutation in <i>CACNA1S</i> caused a conformational change in the protein structure, and the amino acid of this mutation site was highly conserved among different species. <i>SCN4A</i> mutations led to two phenotypes of HypoPP2 and NormoPP. PPPs are autosomal dominant disorders of ion channel dysfunction characterized by episodic flaccid muscle weakness secondary to abnormal sarcolemmal excitability. PPPs are caused by mutations in skeletal muscle calcium channel Ca<sub>V</sub>1.1 gene (<i>CACNA1S</i>), sodium channel Na<sub>V</sub>1.4 gene (<i>SCN4A</i>), and potassium channels Kir2.1, Kir3.4 genes (<i>KCNJ2, KCNJ5</i>), including HypoPP1, HypoPP2, NormoPP, HyperPP, and ATS, which have significant clinical and genetic heterogeneity. Diagnosis is based on the characteristic clinical presentation then confirmed by genetic testing.</p>\",\"PeriodicalId\":72555,\"journal\":{\"name\":\"Channels (Austin, Tex.)\",\"volume\":\" \",\"pages\":\"20-30\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7757828/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Channels (Austin, Tex.)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1080/19336950.2020.1857980\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Channels (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/19336950.2020.1857980","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
The clinical and genetic heterogeneity analysis of five families with primary periodic paralysis.
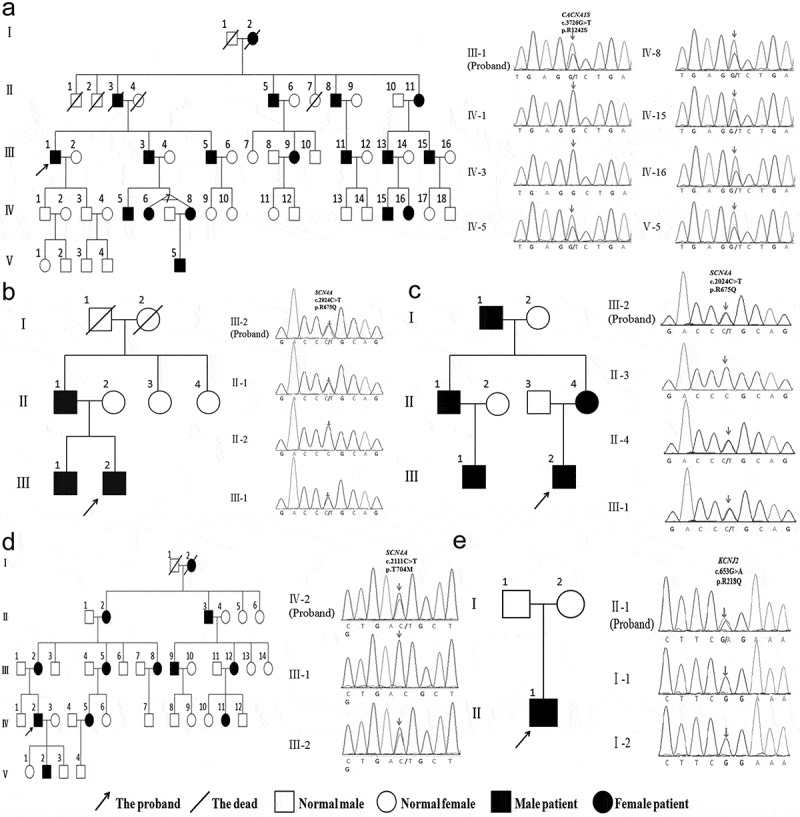
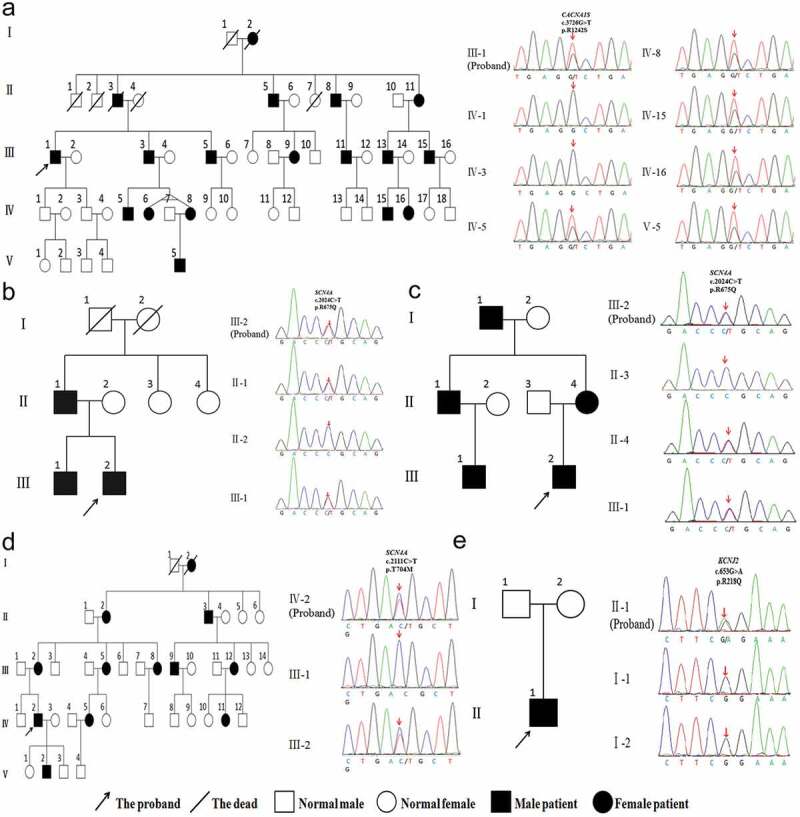
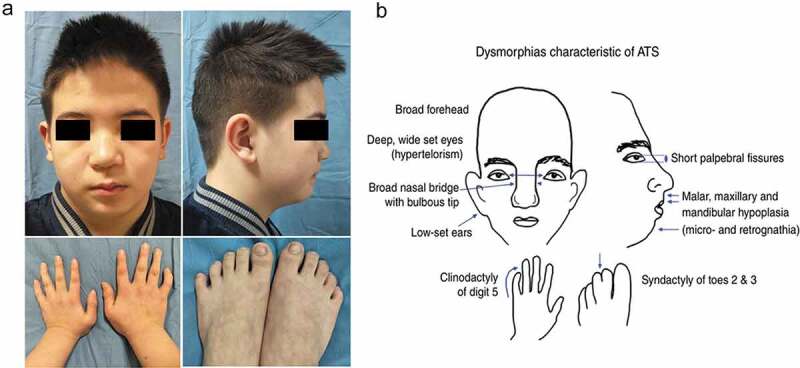
To explore the clinical and genetic characteristics of five families with primary periodic paralysis (PPP). We reviewed clinical manifestations, laboratory results, electrocardiogram, electromyography, muscle biopsy, and genetic analysis from five families with PPP. Five families with PPP included: hypokalemic periodic paralysis type 1 (HypoPP1, CACNA1S, 1/5), hypokalemic periodic paralysis type 2 (HypoPP2, SCN4A, 2/5), normokalemic periodic paralysis (NormoPP, SCN4A, 1/5), and Andersen-Tawil syndrome (ATS, KCNJ2, 1/5). The basic clinical manifestations of five families were consistent with PPP, presenting with paroxysmal muscle weakness, with or without abnormal serum potassium. ATS was accompanied by ventricular arrhythmias, and skeletal and craniofacial anomalies, developing with a permanent fixed myopathy later. The electromyography showed diffuse myopathic discharge, and muscle biopsy showed tubular aggregates. Genetic testing revealed five families with PPP carried CACNA1S (R1242S), SCN4A (R675Q, T704M), and KCNJ2 (R218Q) respectively. The novel heterozygous R1242S mutation in CACNA1S caused a conformational change in the protein structure, and the amino acid of this mutation site was highly conserved among different species. SCN4A mutations led to two phenotypes of HypoPP2 and NormoPP. PPPs are autosomal dominant disorders of ion channel dysfunction characterized by episodic flaccid muscle weakness secondary to abnormal sarcolemmal excitability. PPPs are caused by mutations in skeletal muscle calcium channel CaV1.1 gene (CACNA1S), sodium channel NaV1.4 gene (SCN4A), and potassium channels Kir2.1, Kir3.4 genes (KCNJ2, KCNJ5), including HypoPP1, HypoPP2, NormoPP, HyperPP, and ATS, which have significant clinical and genetic heterogeneity. Diagnosis is based on the characteristic clinical presentation then confirmed by genetic testing.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


