{"title":"细胞朊蛋白在神经退行性疾病病理聚集物的摄取和信号传导中的作用。","authors":"Giuseppe Legname, Carlo Scialò","doi":"10.1080/19336896.2020.1854034","DOIUrl":null,"url":null,"abstract":"<p><p>Neurodegenerative disorders are associated with intra- or extra-cellular deposition of aggregates of misfolded insoluble proteins. These deposits composed of tau, amyloid-β or α-synuclein spread from cell to cell, in a prion-like manner. Novel evidence suggests that the circulating soluble oligomeric species of these misfolded proteins could play a major role in pathology, while insoluble aggregates would represent their protective less toxic counterparts. Recent convincing data support the proposition that the cellular prion protein, PrP<sup>C</sup>, act as a toxicity-inducing receptor for amyloid-β oligomers. As a consequence, several studies focused their investigations to the role played by PrP<sup>C</sup> in binding other protein aggregates, such as tau and α-synuclein, for its possible common role in mediating toxic signalling. The biological relevance of PrP<sup>C</sup> as key ligand and potential mediator of toxicity for multiple proteinaceous aggregated species, prions or PrP<sup>Sc</sup> included, could lead to relevant therapeutic implications. Here we describe the structure of PrP<sup>C</sup> and the proposed interplay with its pathological counterpart PrP<sup>Sc</sup> and then we recapitulate the most recent findings regarding the role of PrP<sup>C</sup> in the interaction with aggregated forms of other neurodegeneration-associated proteins.</p>","PeriodicalId":54585,"journal":{"name":"Prion","volume":" ","pages":"257-270"},"PeriodicalIF":1.6000,"publicationDate":"2020-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1080/19336896.2020.1854034","citationCount":"11","resultStr":"{\"title\":\"On the role of the cellular prion protein in the uptake and signaling of pathological aggregates in neurodegenerative diseases.\",\"authors\":\"Giuseppe Legname, Carlo Scialò\",\"doi\":\"10.1080/19336896.2020.1854034\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Neurodegenerative disorders are associated with intra- or extra-cellular deposition of aggregates of misfolded insoluble proteins. These deposits composed of tau, amyloid-β or α-synuclein spread from cell to cell, in a prion-like manner. Novel evidence suggests that the circulating soluble oligomeric species of these misfolded proteins could play a major role in pathology, while insoluble aggregates would represent their protective less toxic counterparts. Recent convincing data support the proposition that the cellular prion protein, PrP<sup>C</sup>, act as a toxicity-inducing receptor for amyloid-β oligomers. As a consequence, several studies focused their investigations to the role played by PrP<sup>C</sup> in binding other protein aggregates, such as tau and α-synuclein, for its possible common role in mediating toxic signalling. The biological relevance of PrP<sup>C</sup> as key ligand and potential mediator of toxicity for multiple proteinaceous aggregated species, prions or PrP<sup>Sc</sup> included, could lead to relevant therapeutic implications. Here we describe the structure of PrP<sup>C</sup> and the proposed interplay with its pathological counterpart PrP<sup>Sc</sup> and then we recapitulate the most recent findings regarding the role of PrP<sup>C</sup> in the interaction with aggregated forms of other neurodegeneration-associated proteins.</p>\",\"PeriodicalId\":54585,\"journal\":{\"name\":\"Prion\",\"volume\":\" \",\"pages\":\"257-270\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2020-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1080/19336896.2020.1854034\",\"citationCount\":\"11\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Prion\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1080/19336896.2020.1854034\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Prion","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1080/19336896.2020.1854034","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
On the role of the cellular prion protein in the uptake and signaling of pathological aggregates in neurodegenerative diseases.
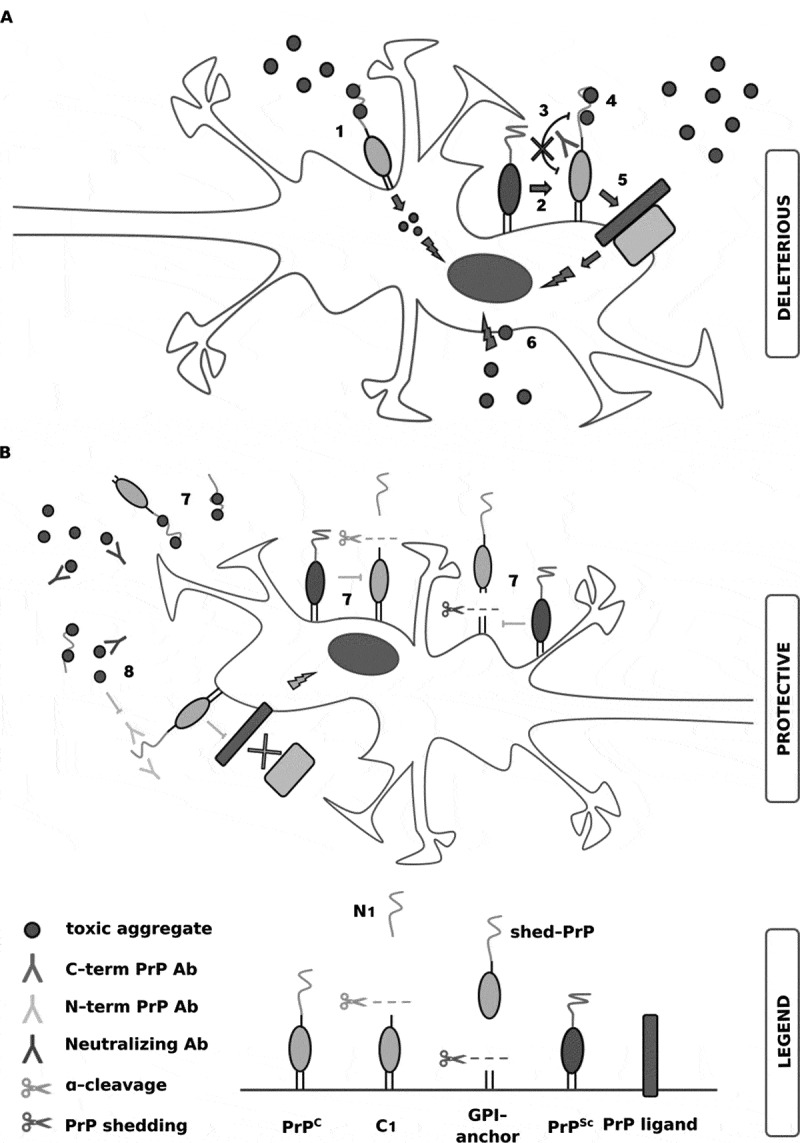
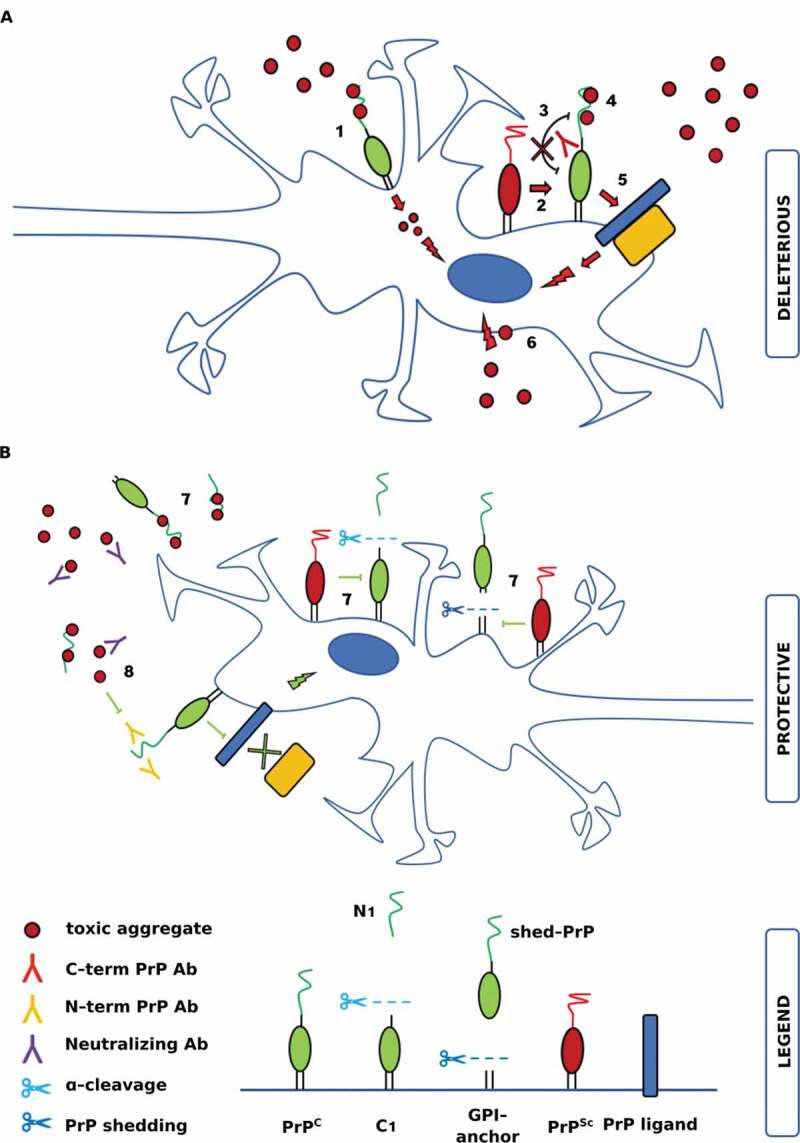
Neurodegenerative disorders are associated with intra- or extra-cellular deposition of aggregates of misfolded insoluble proteins. These deposits composed of tau, amyloid-β or α-synuclein spread from cell to cell, in a prion-like manner. Novel evidence suggests that the circulating soluble oligomeric species of these misfolded proteins could play a major role in pathology, while insoluble aggregates would represent their protective less toxic counterparts. Recent convincing data support the proposition that the cellular prion protein, PrPC, act as a toxicity-inducing receptor for amyloid-β oligomers. As a consequence, several studies focused their investigations to the role played by PrPC in binding other protein aggregates, such as tau and α-synuclein, for its possible common role in mediating toxic signalling. The biological relevance of PrPC as key ligand and potential mediator of toxicity for multiple proteinaceous aggregated species, prions or PrPSc included, could lead to relevant therapeutic implications. Here we describe the structure of PrPC and the proposed interplay with its pathological counterpart PrPSc and then we recapitulate the most recent findings regarding the role of PrPC in the interaction with aggregated forms of other neurodegeneration-associated proteins.
期刊介绍:
Prion is the first international peer-reviewed open access journal to focus exclusively on protein folding and misfolding, protein assembly disorders, protein-based and structural inheritance. The goal is to foster communication and rapid exchange of information through timely publication of important results using traditional as well as electronic formats. The overriding criteria for publication in Prion are originality, scientific merit and general interest.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


