{"title":"基于MALDI质谱法测定的蛋白质动力学深度分析软件的开发。","authors":"Tatsuya Yamamoto, Tohru Yamagaki, Honoo Satake","doi":"10.5702/massspectrometry.S0082","DOIUrl":null,"url":null,"abstract":"<p><p>Hydrogen/deuterium exchange (HDX) coupled with pepsin digestion is useful for rapidly analyzing the kinetic properties of small amounts of protein. However, the analysis of HDX by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) is time-consuming due to a lack of dedicated software. Currently available software programs mainly calculate average mass shifts, even though the isotopic distribution width contains information regarding multiple protein conformations. Moreover, HDX reaction samples are typically composed of peptides that contain various numbers of deuterium atoms, which also hinders the rapid and comprehensive analysis of protein dynamics. We report here on the development of a software program \"Scipas DX\" that can be used to automatically analyze the hydrogen-deuterium isotopic distribution in peaks in HDX spectra and calculate the average number of atoms exchanged, the average deuteration ratio, the abundance ratio for exchanged atoms, and their fitted spectra with a high degree of accuracy within a few minutes. Analysis of the abundance ratio for exchanged atoms of a model protein, adenylate kinase 1, using Scipas DX indicate that the local structure at residues 83-106 and 107-117 are in a slow equilibrium, suggesting that these regions adopt multiple conformations that are involved in the stability and in switching between the active and inactive forms. Furthermore, precise HDX kinetics of the average deuteration ratio both confirmed the known induced conformations of two regions (residues 46-75 and 131-165) that are responsible for ligand binding and verified the novel structural dynamics of residues 107-117 and 166-196 following ligand binding to ligand-binding pockets 1 and 2, respectively. Collectively, these results highlight the usefulness and versatility of Scipas DX in MALDI-MS HDX-based analyses of protein dynamics.</p>","PeriodicalId":18243,"journal":{"name":"Mass spectrometry","volume":"8 2","pages":"S0082"},"PeriodicalIF":0.0000,"publicationDate":"2019-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.5702/massspectrometry.S0082","citationCount":"0","resultStr":"{\"title\":\"Development of Software for the In-Depth Analysis of Protein Dynamics as Determined by MALDI Mass Spectrometry-Based Hydrogen/Deuterium Exchange.\",\"authors\":\"Tatsuya Yamamoto, Tohru Yamagaki, Honoo Satake\",\"doi\":\"10.5702/massspectrometry.S0082\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hydrogen/deuterium exchange (HDX) coupled with pepsin digestion is useful for rapidly analyzing the kinetic properties of small amounts of protein. However, the analysis of HDX by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) is time-consuming due to a lack of dedicated software. Currently available software programs mainly calculate average mass shifts, even though the isotopic distribution width contains information regarding multiple protein conformations. Moreover, HDX reaction samples are typically composed of peptides that contain various numbers of deuterium atoms, which also hinders the rapid and comprehensive analysis of protein dynamics. We report here on the development of a software program \\\"Scipas DX\\\" that can be used to automatically analyze the hydrogen-deuterium isotopic distribution in peaks in HDX spectra and calculate the average number of atoms exchanged, the average deuteration ratio, the abundance ratio for exchanged atoms, and their fitted spectra with a high degree of accuracy within a few minutes. Analysis of the abundance ratio for exchanged atoms of a model protein, adenylate kinase 1, using Scipas DX indicate that the local structure at residues 83-106 and 107-117 are in a slow equilibrium, suggesting that these regions adopt multiple conformations that are involved in the stability and in switching between the active and inactive forms. Furthermore, precise HDX kinetics of the average deuteration ratio both confirmed the known induced conformations of two regions (residues 46-75 and 131-165) that are responsible for ligand binding and verified the novel structural dynamics of residues 107-117 and 166-196 following ligand binding to ligand-binding pockets 1 and 2, respectively. Collectively, these results highlight the usefulness and versatility of Scipas DX in MALDI-MS HDX-based analyses of protein dynamics.</p>\",\"PeriodicalId\":18243,\"journal\":{\"name\":\"Mass spectrometry\",\"volume\":\"8 2\",\"pages\":\"S0082\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2019-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.5702/massspectrometry.S0082\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Mass spectrometry\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5702/massspectrometry.S0082\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2020/2/14 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"Physics and Astronomy\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Mass spectrometry","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5702/massspectrometry.S0082","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/2/14 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"Physics and Astronomy","Score":null,"Total":0}
Development of Software for the In-Depth Analysis of Protein Dynamics as Determined by MALDI Mass Spectrometry-Based Hydrogen/Deuterium Exchange.
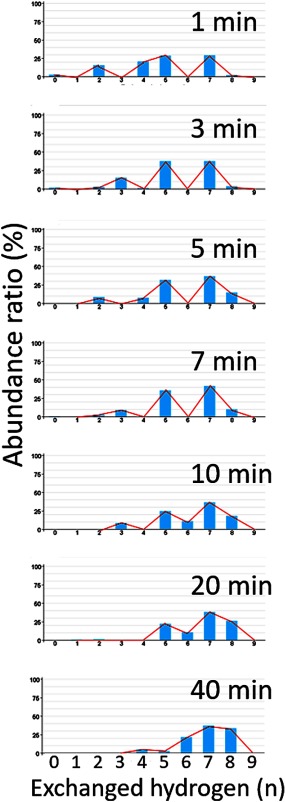
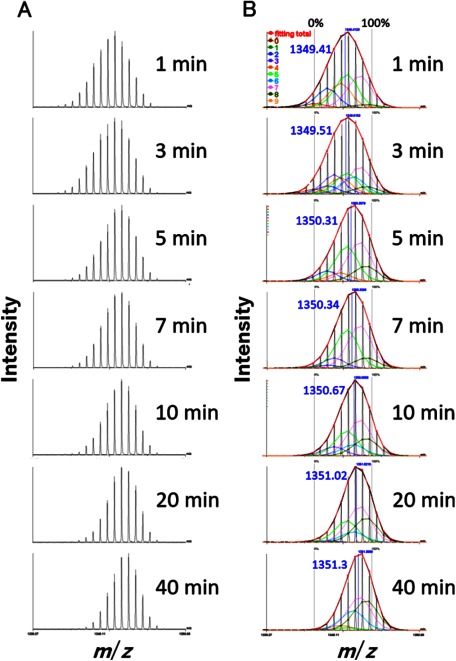
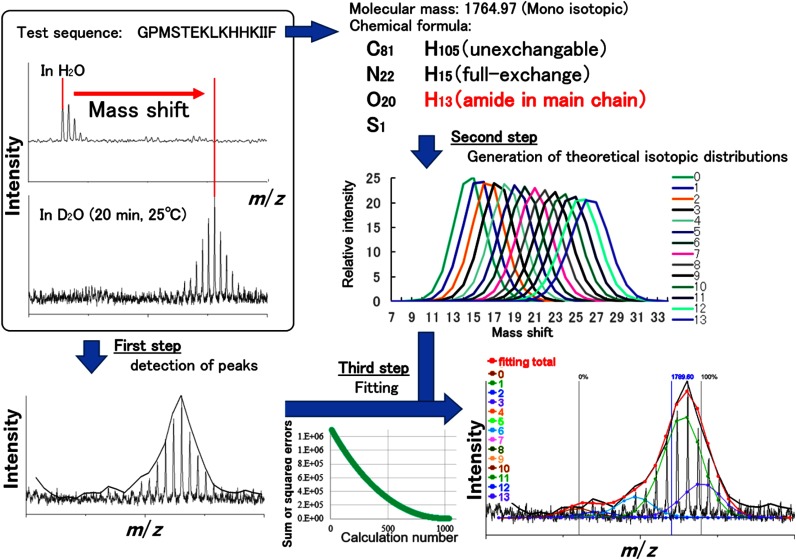
Hydrogen/deuterium exchange (HDX) coupled with pepsin digestion is useful for rapidly analyzing the kinetic properties of small amounts of protein. However, the analysis of HDX by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) is time-consuming due to a lack of dedicated software. Currently available software programs mainly calculate average mass shifts, even though the isotopic distribution width contains information regarding multiple protein conformations. Moreover, HDX reaction samples are typically composed of peptides that contain various numbers of deuterium atoms, which also hinders the rapid and comprehensive analysis of protein dynamics. We report here on the development of a software program "Scipas DX" that can be used to automatically analyze the hydrogen-deuterium isotopic distribution in peaks in HDX spectra and calculate the average number of atoms exchanged, the average deuteration ratio, the abundance ratio for exchanged atoms, and their fitted spectra with a high degree of accuracy within a few minutes. Analysis of the abundance ratio for exchanged atoms of a model protein, adenylate kinase 1, using Scipas DX indicate that the local structure at residues 83-106 and 107-117 are in a slow equilibrium, suggesting that these regions adopt multiple conformations that are involved in the stability and in switching between the active and inactive forms. Furthermore, precise HDX kinetics of the average deuteration ratio both confirmed the known induced conformations of two regions (residues 46-75 and 131-165) that are responsible for ligand binding and verified the novel structural dynamics of residues 107-117 and 166-196 following ligand binding to ligand-binding pockets 1 and 2, respectively. Collectively, these results highlight the usefulness and versatility of Scipas DX in MALDI-MS HDX-based analyses of protein dynamics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


