Kamalika Sen, Arijita Sarkar, Ranjan Kumar Maji, Zhumur Ghosh, Sanjib Gupta and Tapash Chandra Ghosh
{"title":"慢性髓性白血病K562细胞系中人竞争性内源性rna的串扰解译","authors":"Kamalika Sen, Arijita Sarkar, Ranjan Kumar Maji, Zhumur Ghosh, Sanjib Gupta and Tapash Chandra Ghosh","doi":"10.1039/C6MB00568C","DOIUrl":null,"url":null,"abstract":"<p >Chronic myelogenous leukemia (CML) is a myeloproliferative disorder characterized by increased proliferation or abnormal accumulation of granulocytic cell line without the depletion of their capacity to differentiate. A reciprocal chromosomal translocation proceeding to the ‘Philadelphia chromosome', involving the ABL proto-oncogene and BCR gene residing on Chromosome 9 and 22 respectively, is observed to be attributed to CML pathogenesis. Recent studies have been unraveling the crucial role of genomic ‘dark matter’ or the non-coding repertoire in cancer initiation and progression. The intricate cross-talk between competitive endogenous RNAs (ceRNAs) provides a scaffold to systematically functionalize the miRNA response element harboring non-coding RNAs and incorporate them with the protein-coding RNA dimension in complex ceRNA networks. This network of coding and non-coding transcriptome linked by shared miRNAs evidently offers a platform to elucidate the complex regulatory interactions at the post-transcriptional level in human cancers. In this context, analyzing CML, from the perspective of the ceRNA hypothesis, surely craves intensive attention and a comprehensive discussion. Here, we performed RNA-seq data analysis to retrieve Lymphoblastoid and CML coding as well as non-coding repertoire and constructed a ceRNA network for the CML cell line, considering the non-cancer lymphoblastoid cell line as the control. We investigated if any alteration exists in the ceRNA landscape of the transcripts which are exhibiting differential expression across the two cell lines and observed that the major ceRNA regulators vary in cancer network when compared with the Lymphoblastoid network. The top ranked significant functional modules in the ceRNA network display cancer associated attributes and reveal putative regulators in CML pathogenesis.</p>","PeriodicalId":90,"journal":{"name":"Molecular BioSystems","volume":" 12","pages":" 3633-3642"},"PeriodicalIF":3.7430,"publicationDate":"2016-10-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1039/C6MB00568C","citationCount":"4","resultStr":"{\"title\":\"Deciphering the cross-talking of human competitive endogenous RNAs in K562 chronic myelogenous leukemia cell line†\",\"authors\":\"Kamalika Sen, Arijita Sarkar, Ranjan Kumar Maji, Zhumur Ghosh, Sanjib Gupta and Tapash Chandra Ghosh\",\"doi\":\"10.1039/C6MB00568C\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Chronic myelogenous leukemia (CML) is a myeloproliferative disorder characterized by increased proliferation or abnormal accumulation of granulocytic cell line without the depletion of their capacity to differentiate. A reciprocal chromosomal translocation proceeding to the ‘Philadelphia chromosome', involving the ABL proto-oncogene and BCR gene residing on Chromosome 9 and 22 respectively, is observed to be attributed to CML pathogenesis. Recent studies have been unraveling the crucial role of genomic ‘dark matter’ or the non-coding repertoire in cancer initiation and progression. The intricate cross-talk between competitive endogenous RNAs (ceRNAs) provides a scaffold to systematically functionalize the miRNA response element harboring non-coding RNAs and incorporate them with the protein-coding RNA dimension in complex ceRNA networks. This network of coding and non-coding transcriptome linked by shared miRNAs evidently offers a platform to elucidate the complex regulatory interactions at the post-transcriptional level in human cancers. In this context, analyzing CML, from the perspective of the ceRNA hypothesis, surely craves intensive attention and a comprehensive discussion. Here, we performed RNA-seq data analysis to retrieve Lymphoblastoid and CML coding as well as non-coding repertoire and constructed a ceRNA network for the CML cell line, considering the non-cancer lymphoblastoid cell line as the control. We investigated if any alteration exists in the ceRNA landscape of the transcripts which are exhibiting differential expression across the two cell lines and observed that the major ceRNA regulators vary in cancer network when compared with the Lymphoblastoid network. The top ranked significant functional modules in the ceRNA network display cancer associated attributes and reveal putative regulators in CML pathogenesis.</p>\",\"PeriodicalId\":90,\"journal\":{\"name\":\"Molecular BioSystems\",\"volume\":\" 12\",\"pages\":\" 3633-3642\"},\"PeriodicalIF\":3.7430,\"publicationDate\":\"2016-10-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1039/C6MB00568C\",\"citationCount\":\"4\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular BioSystems\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2016/mb/c6mb00568c\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular BioSystems","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2016/mb/c6mb00568c","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Deciphering the cross-talking of human competitive endogenous RNAs in K562 chronic myelogenous leukemia cell line†
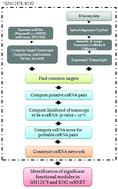
Chronic myelogenous leukemia (CML) is a myeloproliferative disorder characterized by increased proliferation or abnormal accumulation of granulocytic cell line without the depletion of their capacity to differentiate. A reciprocal chromosomal translocation proceeding to the ‘Philadelphia chromosome', involving the ABL proto-oncogene and BCR gene residing on Chromosome 9 and 22 respectively, is observed to be attributed to CML pathogenesis. Recent studies have been unraveling the crucial role of genomic ‘dark matter’ or the non-coding repertoire in cancer initiation and progression. The intricate cross-talk between competitive endogenous RNAs (ceRNAs) provides a scaffold to systematically functionalize the miRNA response element harboring non-coding RNAs and incorporate them with the protein-coding RNA dimension in complex ceRNA networks. This network of coding and non-coding transcriptome linked by shared miRNAs evidently offers a platform to elucidate the complex regulatory interactions at the post-transcriptional level in human cancers. In this context, analyzing CML, from the perspective of the ceRNA hypothesis, surely craves intensive attention and a comprehensive discussion. Here, we performed RNA-seq data analysis to retrieve Lymphoblastoid and CML coding as well as non-coding repertoire and constructed a ceRNA network for the CML cell line, considering the non-cancer lymphoblastoid cell line as the control. We investigated if any alteration exists in the ceRNA landscape of the transcripts which are exhibiting differential expression across the two cell lines and observed that the major ceRNA regulators vary in cancer network when compared with the Lymphoblastoid network. The top ranked significant functional modules in the ceRNA network display cancer associated attributes and reveal putative regulators in CML pathogenesis.
期刊介绍:
Molecular Omics publishes molecular level experimental and bioinformatics research in the -omics sciences, including genomics, proteomics, transcriptomics and metabolomics. We will also welcome multidisciplinary papers presenting studies combining different types of omics, or the interface of omics and other fields such as systems biology or chemical biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


