Xiaolin Ni, Chenxi Jin, Yan Jiang, Ou Wang, Mei Li, Xiaoping Xing, Weibo Xia
{"title":"伴PLOD1基因突变的中国后凸性埃勒-丹洛斯综合征首例报道。","authors":"Xiaolin Ni, Chenxi Jin, Yan Jiang, Ou Wang, Mei Li, Xiaoping Xing, Weibo Xia","doi":"10.1186/s12881-020-01154-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Kyphoscoliotic Ehlers-Danlos syndrome (kEDS) is a rare autosomal recessive connective tissue disorder characterized by progressive kyphoscoliosis, congenital muscular hypotonia, marked joint hypermobility, and severe skin hyperextensibility and fragility. Deficiency of lysyl hydroxylase 1 (LH1) due to mutations of PLOD1 (procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1) gene has been identified as the pathogenic cause of kEDS (kEDS-PLOD1). Up to now, kEDS-PLOD1 has not been reported among Chinese population.</p><p><strong>Case presentation: </strong>A 17-year-old Chinese male patient presenting with hypotonia, joint hypermobility and scoliosis was referred to our hospital. After birth, he was found to have severe hypotonia leading to delayed motor development. Subsequently, joint hypermobility, kyphoscoliosis and amblyopia were found. Inguinal hernia was found at age 5 years and closed by surgery. At the same time, he presented with hyperextensible and bruisable velvety skin with widened atrophic scarring after minor trauma. Dislocation of elbow joint was noted at age of 6 years. Orthopedic surgery for correction of kyphoscoliosis was performed at age 10 years. His family history was unremarkable. Physical examination revealed elevated blood pressure. Slight facial dysmorphologies including high palate, epicanthal folds, and down-slanting palpebral fissures were found. He also had blue sclerae with normal hearing. X-rays revealed severe degree of scoliosis and osteopenia. The Echocardiography findings were normal. Laboratory examination revealed a slightly elevated bone turnover. Based on the clinical manifestations presented by our patient, kEDS was suspected. Genetic analysis revealed a novel homozygous missense mutation of PLOD1 (c.1697 G > A, p.C566Y), confirming the diagnosis of kEDS-PLOD1. The patient was treated with alfacalcidol and nifedipine. Improved physical strength and normal blood pressure were reported after 12-month follow-up.</p><p><strong>Conclusions: </strong>This is the first case of kEDS-PLOD1 of Chinese origin. We identified one novel mutation of PLOD1, extending the mutation spectrum of PLOD1. Diagnosis of kEDS-PLOD1 should be considered in patients with congenital hypotonia, progressive kyphoscoliosis, joint hypermobility, and skin hyperextensibility and confirmed by mutation analysis of PLOD1.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"214"},"PeriodicalIF":0.0000,"publicationDate":"2020-10-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01154-3","citationCount":"3","resultStr":"{\"title\":\"The first case report of Kyphoscoliotic Ehlers-Danlos syndrome of chinese origin with a novel PLOD1 gene mutation.\",\"authors\":\"Xiaolin Ni, Chenxi Jin, Yan Jiang, Ou Wang, Mei Li, Xiaoping Xing, Weibo Xia\",\"doi\":\"10.1186/s12881-020-01154-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Kyphoscoliotic Ehlers-Danlos syndrome (kEDS) is a rare autosomal recessive connective tissue disorder characterized by progressive kyphoscoliosis, congenital muscular hypotonia, marked joint hypermobility, and severe skin hyperextensibility and fragility. Deficiency of lysyl hydroxylase 1 (LH1) due to mutations of PLOD1 (procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1) gene has been identified as the pathogenic cause of kEDS (kEDS-PLOD1). Up to now, kEDS-PLOD1 has not been reported among Chinese population.</p><p><strong>Case presentation: </strong>A 17-year-old Chinese male patient presenting with hypotonia, joint hypermobility and scoliosis was referred to our hospital. After birth, he was found to have severe hypotonia leading to delayed motor development. Subsequently, joint hypermobility, kyphoscoliosis and amblyopia were found. Inguinal hernia was found at age 5 years and closed by surgery. At the same time, he presented with hyperextensible and bruisable velvety skin with widened atrophic scarring after minor trauma. Dislocation of elbow joint was noted at age of 6 years. Orthopedic surgery for correction of kyphoscoliosis was performed at age 10 years. His family history was unremarkable. Physical examination revealed elevated blood pressure. Slight facial dysmorphologies including high palate, epicanthal folds, and down-slanting palpebral fissures were found. He also had blue sclerae with normal hearing. X-rays revealed severe degree of scoliosis and osteopenia. The Echocardiography findings were normal. Laboratory examination revealed a slightly elevated bone turnover. Based on the clinical manifestations presented by our patient, kEDS was suspected. Genetic analysis revealed a novel homozygous missense mutation of PLOD1 (c.1697 G > A, p.C566Y), confirming the diagnosis of kEDS-PLOD1. The patient was treated with alfacalcidol and nifedipine. Improved physical strength and normal blood pressure were reported after 12-month follow-up.</p><p><strong>Conclusions: </strong>This is the first case of kEDS-PLOD1 of Chinese origin. We identified one novel mutation of PLOD1, extending the mutation spectrum of PLOD1. Diagnosis of kEDS-PLOD1 should be considered in patients with congenital hypotonia, progressive kyphoscoliosis, joint hypermobility, and skin hyperextensibility and confirmed by mutation analysis of PLOD1.</p>\",\"PeriodicalId\":9015,\"journal\":{\"name\":\"BMC Medical Genetics\",\"volume\":\" \",\"pages\":\"214\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-10-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12881-020-01154-3\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12881-020-01154-3\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01154-3","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
The first case report of Kyphoscoliotic Ehlers-Danlos syndrome of chinese origin with a novel PLOD1 gene mutation.
Background: Kyphoscoliotic Ehlers-Danlos syndrome (kEDS) is a rare autosomal recessive connective tissue disorder characterized by progressive kyphoscoliosis, congenital muscular hypotonia, marked joint hypermobility, and severe skin hyperextensibility and fragility. Deficiency of lysyl hydroxylase 1 (LH1) due to mutations of PLOD1 (procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1) gene has been identified as the pathogenic cause of kEDS (kEDS-PLOD1). Up to now, kEDS-PLOD1 has not been reported among Chinese population.
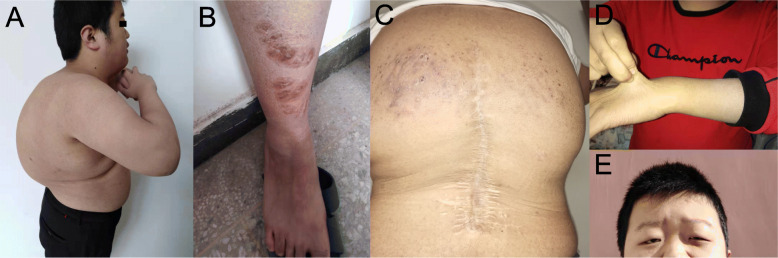
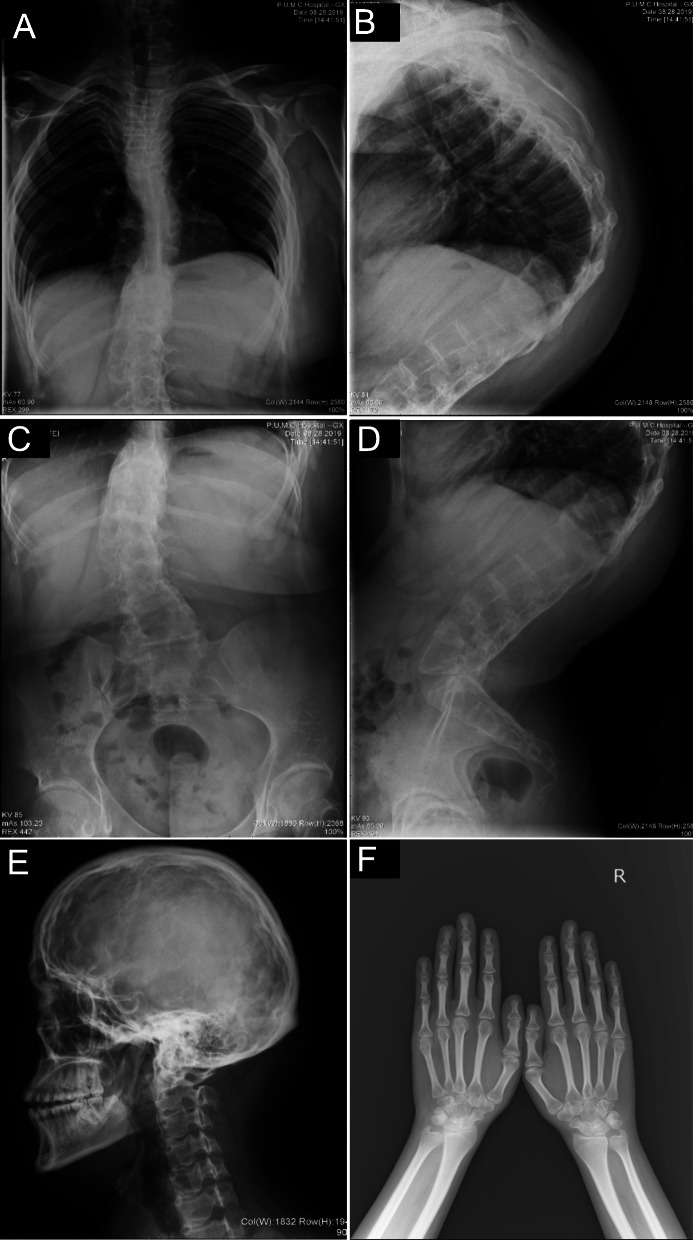
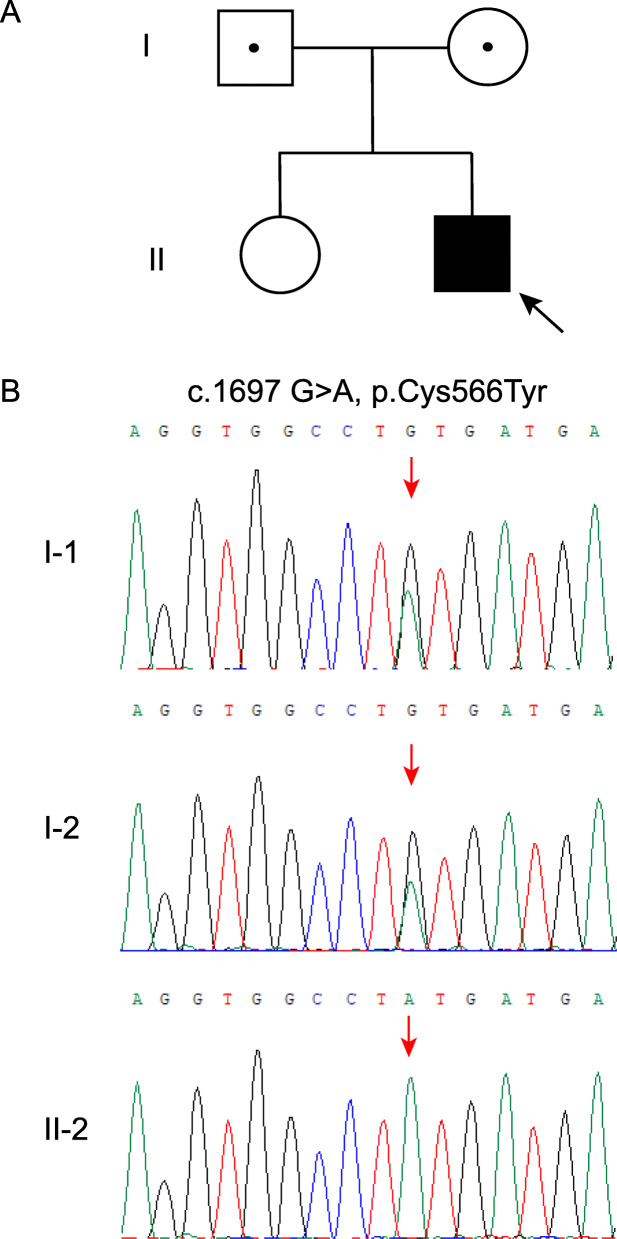
Case presentation: A 17-year-old Chinese male patient presenting with hypotonia, joint hypermobility and scoliosis was referred to our hospital. After birth, he was found to have severe hypotonia leading to delayed motor development. Subsequently, joint hypermobility, kyphoscoliosis and amblyopia were found. Inguinal hernia was found at age 5 years and closed by surgery. At the same time, he presented with hyperextensible and bruisable velvety skin with widened atrophic scarring after minor trauma. Dislocation of elbow joint was noted at age of 6 years. Orthopedic surgery for correction of kyphoscoliosis was performed at age 10 years. His family history was unremarkable. Physical examination revealed elevated blood pressure. Slight facial dysmorphologies including high palate, epicanthal folds, and down-slanting palpebral fissures were found. He also had blue sclerae with normal hearing. X-rays revealed severe degree of scoliosis and osteopenia. The Echocardiography findings were normal. Laboratory examination revealed a slightly elevated bone turnover. Based on the clinical manifestations presented by our patient, kEDS was suspected. Genetic analysis revealed a novel homozygous missense mutation of PLOD1 (c.1697 G > A, p.C566Y), confirming the diagnosis of kEDS-PLOD1. The patient was treated with alfacalcidol and nifedipine. Improved physical strength and normal blood pressure were reported after 12-month follow-up.
Conclusions: This is the first case of kEDS-PLOD1 of Chinese origin. We identified one novel mutation of PLOD1, extending the mutation spectrum of PLOD1. Diagnosis of kEDS-PLOD1 should be considered in patients with congenital hypotonia, progressive kyphoscoliosis, joint hypermobility, and skin hyperextensibility and confirmed by mutation analysis of PLOD1.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:



