{"title":"诊断为不完全阿拉吉尔综合征的法洛四联症成人患者的腹膜透析。","authors":"Malgorzata Ponikowska, Agnieszka Pollak, Ewa Kotwica-Strzalek, Dorota Brodowska-Kania, Magdalena Mosakowska, Rafal Ploski, Stanislaw Niemczyk","doi":"10.1186/s12881-020-01134-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Alagille syndrome is an autosomal dominant disorder usually caused by pathogenic variants of the JAG1 gene. In the past, cholestasis was a condition sine qua non for diagnosis of the syndrome. However, recent advancements in genetic testing have revealed that clinical presentations vary from lack of symptoms, to multiorgan involvement. Tetralogy of Fallot, the most frequent complex congenital heart defect in Alagille Syndrome, very rarely leads to renal failure requiring dialysis - there are only single reports of such cases in the literature, with none of them in Alagille Syndrome.</p><p><strong>Case presentation: </strong>A 41-year-old woman suffering from cyanosis, dyspnea and plethora was admitted to the hospital. The patient suffered from chronic kidney disease and tetralogy of Fallot and had been treated palliatively with Blalock-Taussig shunts in the past; at admission, only minimal flow through the left shunt was preserved. These symptoms, together with impaired mental status and dysmorphic facial features, led to extensive clinical and genetic testing including whole exome sequencing. A previously unknown missense variant c.587G > A within the JAG1 gene was identified. As there were no signs of cholestasis, and subclinical liver involvement was only suggested by elevated alkaline phosphatase levels, the patient was diagnosed with incomplete Alagille Syndrome. End-stage renal disease required introduction of renal replacement therapy. Continuous ambulatory peritoneal dialysis was chosen and the patient's quality of life significantly increased. However, after refusal of further treatment, the patient died at the age of 45.</p><p><strong>Conclusions: </strong>Tetralogy of Fallot should always urge clinicians to evaluate for Alagille Syndrome and offer patients early nephrological care. Although tetralogy of Fallot rarely leads to end-stage renal disease requiring dialysis, if treated palliatively and combined with renal dysplasia (typical of Alagille Syndrome), it can result in severe renal failure as in the presented case. There is no standard treatment for such cases, but based on our experience, peritoneal dialysis is worth consideration. Finally, clinical criteria for the diagnosis of Alagille Syndrome require revision. Previously, diagnosis was based on cholestasis - however, cardiovascular anomalies are found to be more prevalent. Furthermore, the criteria do not include renal impairment, which is also common.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"195"},"PeriodicalIF":0.0000,"publicationDate":"2020-10-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01134-7","citationCount":"1","resultStr":"{\"title\":\"Peritoneal dialysis in an adult patient with tetralogy of Fallot diagnosed with incomplete Alagille syndrome.\",\"authors\":\"Malgorzata Ponikowska, Agnieszka Pollak, Ewa Kotwica-Strzalek, Dorota Brodowska-Kania, Magdalena Mosakowska, Rafal Ploski, Stanislaw Niemczyk\",\"doi\":\"10.1186/s12881-020-01134-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Alagille syndrome is an autosomal dominant disorder usually caused by pathogenic variants of the JAG1 gene. In the past, cholestasis was a condition sine qua non for diagnosis of the syndrome. However, recent advancements in genetic testing have revealed that clinical presentations vary from lack of symptoms, to multiorgan involvement. Tetralogy of Fallot, the most frequent complex congenital heart defect in Alagille Syndrome, very rarely leads to renal failure requiring dialysis - there are only single reports of such cases in the literature, with none of them in Alagille Syndrome.</p><p><strong>Case presentation: </strong>A 41-year-old woman suffering from cyanosis, dyspnea and plethora was admitted to the hospital. The patient suffered from chronic kidney disease and tetralogy of Fallot and had been treated palliatively with Blalock-Taussig shunts in the past; at admission, only minimal flow through the left shunt was preserved. These symptoms, together with impaired mental status and dysmorphic facial features, led to extensive clinical and genetic testing including whole exome sequencing. A previously unknown missense variant c.587G > A within the JAG1 gene was identified. As there were no signs of cholestasis, and subclinical liver involvement was only suggested by elevated alkaline phosphatase levels, the patient was diagnosed with incomplete Alagille Syndrome. End-stage renal disease required introduction of renal replacement therapy. Continuous ambulatory peritoneal dialysis was chosen and the patient's quality of life significantly increased. However, after refusal of further treatment, the patient died at the age of 45.</p><p><strong>Conclusions: </strong>Tetralogy of Fallot should always urge clinicians to evaluate for Alagille Syndrome and offer patients early nephrological care. Although tetralogy of Fallot rarely leads to end-stage renal disease requiring dialysis, if treated palliatively and combined with renal dysplasia (typical of Alagille Syndrome), it can result in severe renal failure as in the presented case. There is no standard treatment for such cases, but based on our experience, peritoneal dialysis is worth consideration. Finally, clinical criteria for the diagnosis of Alagille Syndrome require revision. Previously, diagnosis was based on cholestasis - however, cardiovascular anomalies are found to be more prevalent. Furthermore, the criteria do not include renal impairment, which is also common.</p>\",\"PeriodicalId\":9015,\"journal\":{\"name\":\"BMC Medical Genetics\",\"volume\":\" \",\"pages\":\"195\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-10-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12881-020-01134-7\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12881-020-01134-7\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01134-7","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 1
摘要
背景:Alagille综合征是一种常染色体显性遗传病,通常由JAG1基因的致病性变异引起。在过去,胆汁淤积是诊断该综合征的必要条件。然而,基因检测的最新进展表明,临床表现各不相同,从缺乏症状到多器官受累。法洛四联症(Fallot Tetralogy of Fallot)是Alagille综合征中最常见的复杂先天性心脏缺陷,很少导致需要透析的肾衰竭——文献中只有单一的此类病例报道,没有一例发生在Alagille综合征中。病例介绍:一名41岁女性因紫绀、呼吸困难和呼吸过多而入院。患者患有慢性肾脏疾病和法洛四联症,过去曾用Blalock-Taussig分流术姑息治疗;入院时,只有极少的血流通过左侧分流。这些症状,加上精神状态受损和面部畸形,导致广泛的临床和基因检测,包括全外显子组测序。在JAG1基因中发现了一种以前未知的错义变异c.587G > A。由于没有胆汁淤积的迹象,并且仅通过碱性磷酸酶水平升高提示亚临床肝脏受累,因此诊断为不完全Alagille综合征。终末期肾病需要引入肾脏替代治疗。选择持续腹膜透析,患者的生活质量明显提高。然而,在拒绝进一步治疗后,患者在45岁时死亡。结论:法洛四联症应始终督促临床医师对Alagille综合征进行评估,并给予患者早期肾病护理。虽然法洛四联症很少导致需要透析的终末期肾脏疾病,但如果姑息治疗并合并肾脏发育不良(典型的Alagille综合征),就会导致严重的肾功能衰竭,正如本病例所示。这种情况没有标准的治疗方法,但根据我们的经验,腹膜透析是值得考虑的。最后,诊断Alagille综合征的临床标准需要修订。以前,诊断是基于胆汁淤积-然而,发现心血管异常更为普遍。此外,标准不包括肾脏损害,这也是常见的。
Peritoneal dialysis in an adult patient with tetralogy of Fallot diagnosed with incomplete Alagille syndrome.
Background: Alagille syndrome is an autosomal dominant disorder usually caused by pathogenic variants of the JAG1 gene. In the past, cholestasis was a condition sine qua non for diagnosis of the syndrome. However, recent advancements in genetic testing have revealed that clinical presentations vary from lack of symptoms, to multiorgan involvement. Tetralogy of Fallot, the most frequent complex congenital heart defect in Alagille Syndrome, very rarely leads to renal failure requiring dialysis - there are only single reports of such cases in the literature, with none of them in Alagille Syndrome.
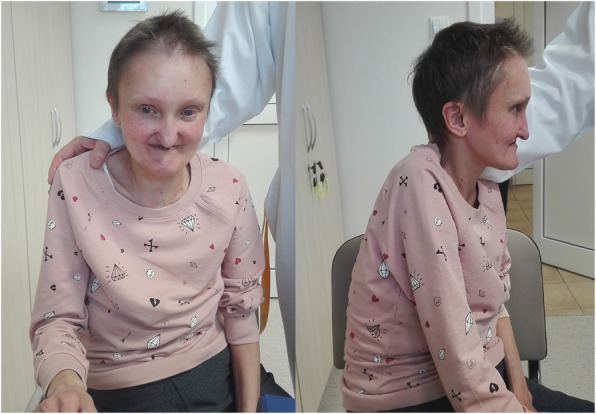
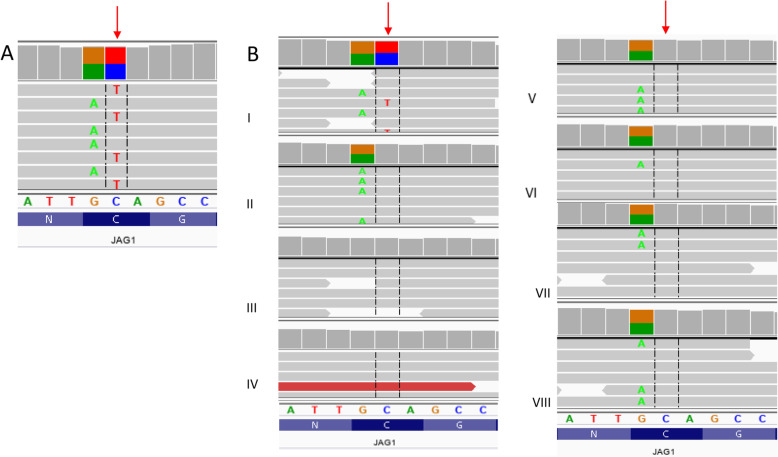
Case presentation: A 41-year-old woman suffering from cyanosis, dyspnea and plethora was admitted to the hospital. The patient suffered from chronic kidney disease and tetralogy of Fallot and had been treated palliatively with Blalock-Taussig shunts in the past; at admission, only minimal flow through the left shunt was preserved. These symptoms, together with impaired mental status and dysmorphic facial features, led to extensive clinical and genetic testing including whole exome sequencing. A previously unknown missense variant c.587G > A within the JAG1 gene was identified. As there were no signs of cholestasis, and subclinical liver involvement was only suggested by elevated alkaline phosphatase levels, the patient was diagnosed with incomplete Alagille Syndrome. End-stage renal disease required introduction of renal replacement therapy. Continuous ambulatory peritoneal dialysis was chosen and the patient's quality of life significantly increased. However, after refusal of further treatment, the patient died at the age of 45.
Conclusions: Tetralogy of Fallot should always urge clinicians to evaluate for Alagille Syndrome and offer patients early nephrological care. Although tetralogy of Fallot rarely leads to end-stage renal disease requiring dialysis, if treated palliatively and combined with renal dysplasia (typical of Alagille Syndrome), it can result in severe renal failure as in the presented case. There is no standard treatment for such cases, but based on our experience, peritoneal dialysis is worth consideration. Finally, clinical criteria for the diagnosis of Alagille Syndrome require revision. Previously, diagnosis was based on cholestasis - however, cardiovascular anomalies are found to be more prevalent. Furthermore, the criteria do not include renal impairment, which is also common.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


