Andrew Currin, Neil Swainston, Mark S Dunstan, Adrian J Jervis, Paul Mulherin, Christopher J Robinson, Sandra Taylor, Pablo Carbonell, Katherine A Hollywood, Cunyu Yan, Eriko Takano, Nigel S Scrutton, Rainer Breitling
{"title":"高度复用、快速准确的纳米孔测序,用于验证合成 DNA 构建和序列文库。","authors":"Andrew Currin, Neil Swainston, Mark S Dunstan, Adrian J Jervis, Paul Mulherin, Christopher J Robinson, Sandra Taylor, Pablo Carbonell, Katherine A Hollywood, Cunyu Yan, Eriko Takano, Nigel S Scrutton, Rainer Breitling","doi":"10.1093/synbio/ysz025","DOIUrl":null,"url":null,"abstract":"<p><p>Synthetic biology utilizes the Design-Build-Test-Learn pipeline for the engineering of biological systems. Typically, this requires the construction of specifically designed, large and complex DNA assemblies. The availability of cheap DNA synthesis and automation enables high-throughput assembly approaches, which generates a heavy demand for DNA sequencing to verify correctly assembled constructs. Next-generation sequencing is ideally positioned to perform this task, however with expensive hardware costs and bespoke data analysis requirements few laboratories utilize this technology in-house. Here a workflow for highly multiplexed sequencing is presented, capable of fast and accurate sequence verification of DNA assemblies using nanopore technology. A novel sample barcoding system using polymerase chain reaction is introduced, and sequencing data are analyzed through a bespoke analysis algorithm. Crucially, this algorithm overcomes the problem of high-error rate nanopore data (which typically prevents identification of single nucleotide variants) through statistical analysis of strand bias, permitting accurate sequence analysis with single-base resolution. As an example, 576 constructs (6 × 96 well plates) were processed in a single workflow in 72 h (from <i>Escherichia coli</i> colonies to analyzed data). Given our procedure's low hardware costs and highly multiplexed capability, this provides cost-effective access to powerful DNA sequencing for any laboratory, with applications beyond synthetic biology including directed evolution, single nucleotide polymorphism analysis and gene synthesis.</p>","PeriodicalId":74902,"journal":{"name":"Synthetic biology (Oxford, England)","volume":"4 1","pages":"ysz025"},"PeriodicalIF":2.6000,"publicationDate":"2019-10-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7445882/pdf/","citationCount":"0","resultStr":"{\"title\":\"Highly multiplexed, fast and accurate nanopore sequencing for verification of synthetic DNA constructs and sequence libraries.\",\"authors\":\"Andrew Currin, Neil Swainston, Mark S Dunstan, Adrian J Jervis, Paul Mulherin, Christopher J Robinson, Sandra Taylor, Pablo Carbonell, Katherine A Hollywood, Cunyu Yan, Eriko Takano, Nigel S Scrutton, Rainer Breitling\",\"doi\":\"10.1093/synbio/ysz025\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Synthetic biology utilizes the Design-Build-Test-Learn pipeline for the engineering of biological systems. Typically, this requires the construction of specifically designed, large and complex DNA assemblies. The availability of cheap DNA synthesis and automation enables high-throughput assembly approaches, which generates a heavy demand for DNA sequencing to verify correctly assembled constructs. Next-generation sequencing is ideally positioned to perform this task, however with expensive hardware costs and bespoke data analysis requirements few laboratories utilize this technology in-house. Here a workflow for highly multiplexed sequencing is presented, capable of fast and accurate sequence verification of DNA assemblies using nanopore technology. A novel sample barcoding system using polymerase chain reaction is introduced, and sequencing data are analyzed through a bespoke analysis algorithm. Crucially, this algorithm overcomes the problem of high-error rate nanopore data (which typically prevents identification of single nucleotide variants) through statistical analysis of strand bias, permitting accurate sequence analysis with single-base resolution. As an example, 576 constructs (6 × 96 well plates) were processed in a single workflow in 72 h (from <i>Escherichia coli</i> colonies to analyzed data). Given our procedure's low hardware costs and highly multiplexed capability, this provides cost-effective access to powerful DNA sequencing for any laboratory, with applications beyond synthetic biology including directed evolution, single nucleotide polymorphism analysis and gene synthesis.</p>\",\"PeriodicalId\":74902,\"journal\":{\"name\":\"Synthetic biology (Oxford, England)\",\"volume\":\"4 1\",\"pages\":\"ysz025\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2019-10-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7445882/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Synthetic biology (Oxford, England)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/synbio/ysz025\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2019/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Synthetic biology (Oxford, England)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/synbio/ysz025","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
摘要
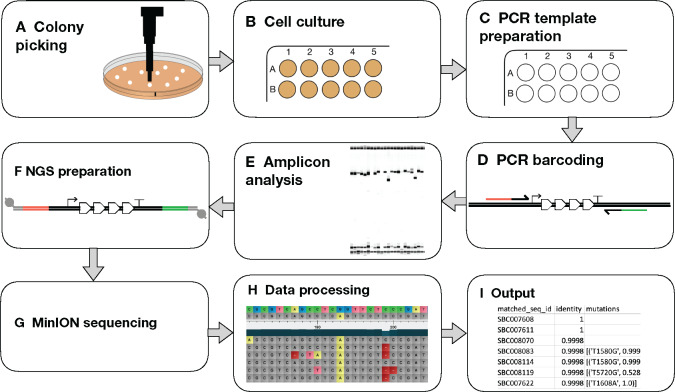
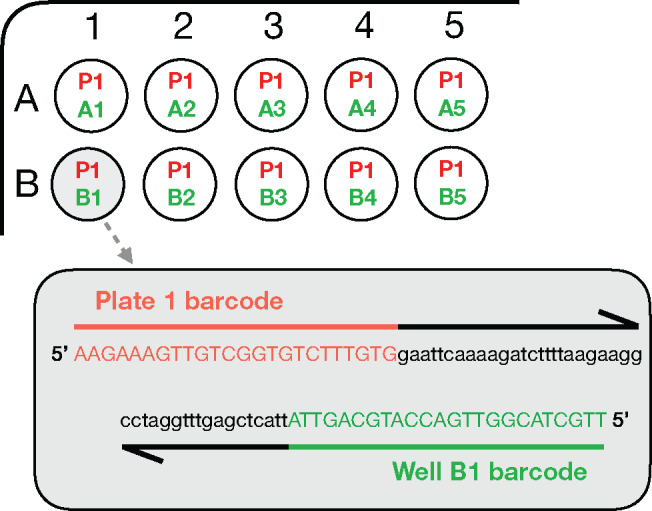
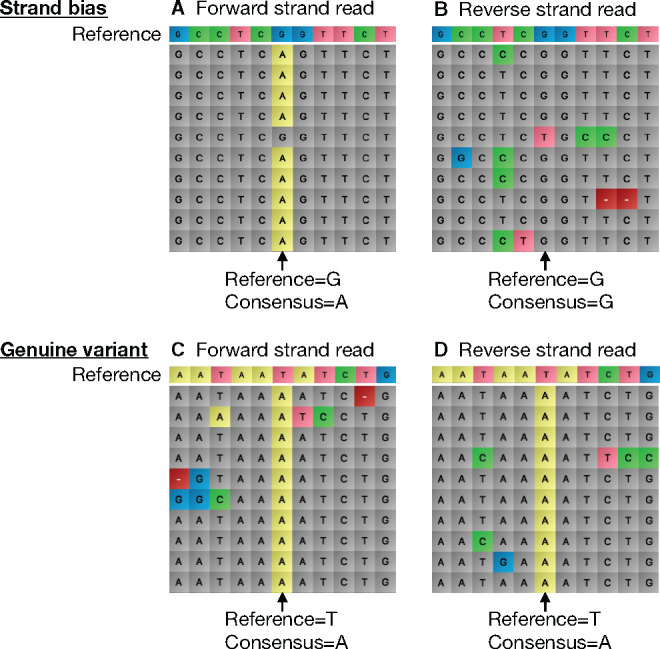
合成生物学利用 "设计-构建-测试-学习 "管道来设计生物系统。通常,这需要构建专门设计的大型复杂 DNA 组合。廉价 DNA 合成技术和自动化技术的出现使高通量组装方法成为可能,从而产生了对 DNA 测序的大量需求,以验证正确组装的构建体。下一代测序技术是完成这项任务的理想选择,但由于硬件成本昂贵,而且需要定制数据分析,因此很少有实验室在内部使用这项技术。本文介绍了一种高度复用测序的工作流程,它能利用纳米孔技术对 DNA 组装进行快速准确的序列验证。介绍了使用聚合酶链反应的新型样品条形码系统,并通过定制的分析算法对测序数据进行分析。最重要的是,该算法通过对链偏差的统计分析,克服了高误差率纳米孔数据的问题(这通常会阻碍对单核苷酸变异的识别),从而实现了单碱基分辨率的精确序列分析。例如,在一个工作流程中处理 576 个构建体(6 × 96 孔板)只需 72 小时(从大肠杆菌菌落到分析数据)。鉴于我们的程序硬件成本低、复用能力强,这为任何实验室提供了具有成本效益的强大 DNA 测序能力,其应用范围超出了合成生物学,包括定向进化、单核苷酸多态性分析和基因合成。
Highly multiplexed, fast and accurate nanopore sequencing for verification of synthetic DNA constructs and sequence libraries.
Synthetic biology utilizes the Design-Build-Test-Learn pipeline for the engineering of biological systems. Typically, this requires the construction of specifically designed, large and complex DNA assemblies. The availability of cheap DNA synthesis and automation enables high-throughput assembly approaches, which generates a heavy demand for DNA sequencing to verify correctly assembled constructs. Next-generation sequencing is ideally positioned to perform this task, however with expensive hardware costs and bespoke data analysis requirements few laboratories utilize this technology in-house. Here a workflow for highly multiplexed sequencing is presented, capable of fast and accurate sequence verification of DNA assemblies using nanopore technology. A novel sample barcoding system using polymerase chain reaction is introduced, and sequencing data are analyzed through a bespoke analysis algorithm. Crucially, this algorithm overcomes the problem of high-error rate nanopore data (which typically prevents identification of single nucleotide variants) through statistical analysis of strand bias, permitting accurate sequence analysis with single-base resolution. As an example, 576 constructs (6 × 96 well plates) were processed in a single workflow in 72 h (from Escherichia coli colonies to analyzed data). Given our procedure's low hardware costs and highly multiplexed capability, this provides cost-effective access to powerful DNA sequencing for any laboratory, with applications beyond synthetic biology including directed evolution, single nucleotide polymorphism analysis and gene synthesis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


