Dipendra C Sengupta, Matthew D Hill, Kevin R Benton, Hirendra N Banerjee
{"title":"通过混沌博弈表示法研究电晕病毒的相似性。","authors":"Dipendra C Sengupta, Matthew D Hill, Kevin R Benton, Hirendra N Banerjee","doi":"10.4236/cmb.2020.103004","DOIUrl":null,"url":null,"abstract":"<p><p>The novel coronavirus (SARS-COV-2) is generally referred to as Covid-19 virus has spread to 213 countries with nearly 7 million confirmed cases and nearly 400,000 deaths. Such major outbreaks demand classification and origin of the virus genomic sequence, for planning, containment, and treatment. Motivated by the above need, we report two alignment-free methods combing with CGR to perform clustering analysis and create a phylogenetic tree based on it. To each DNA sequence we associate a matrix then define distance between two DNA sequences to be the distance between their associated matrix. These methods are being used for phylogenetic analysis of coronavirus sequences. Our approach provides a powerful tool for analyzing and annotating genomes and their phylogenetic relationships. We also compare our tool to ClustalX algorithm which is one of the most popular alignment methods. Our alignment-free methods are shown to be capable of finding closest genetic relatives of coronaviruses.</p>","PeriodicalId":70839,"journal":{"name":"计算分子生物学(英文)","volume":null,"pages":null},"PeriodicalIF":0.0000,"publicationDate":"2020-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7497811/pdf/","citationCount":"0","resultStr":"{\"title\":\"Similarity Studies of Corona Viruses through Chaos Game Representation.\",\"authors\":\"Dipendra C Sengupta, Matthew D Hill, Kevin R Benton, Hirendra N Banerjee\",\"doi\":\"10.4236/cmb.2020.103004\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The novel coronavirus (SARS-COV-2) is generally referred to as Covid-19 virus has spread to 213 countries with nearly 7 million confirmed cases and nearly 400,000 deaths. Such major outbreaks demand classification and origin of the virus genomic sequence, for planning, containment, and treatment. Motivated by the above need, we report two alignment-free methods combing with CGR to perform clustering analysis and create a phylogenetic tree based on it. To each DNA sequence we associate a matrix then define distance between two DNA sequences to be the distance between their associated matrix. These methods are being used for phylogenetic analysis of coronavirus sequences. Our approach provides a powerful tool for analyzing and annotating genomes and their phylogenetic relationships. We also compare our tool to ClustalX algorithm which is one of the most popular alignment methods. Our alignment-free methods are shown to be capable of finding closest genetic relatives of coronaviruses.</p>\",\"PeriodicalId\":70839,\"journal\":{\"name\":\"计算分子生物学(英文)\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7497811/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"计算分子生物学(英文)\",\"FirstCategoryId\":\"1089\",\"ListUrlMain\":\"https://doi.org/10.4236/cmb.2020.103004\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"计算分子生物学(英文)","FirstCategoryId":"1089","ListUrlMain":"https://doi.org/10.4236/cmb.2020.103004","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
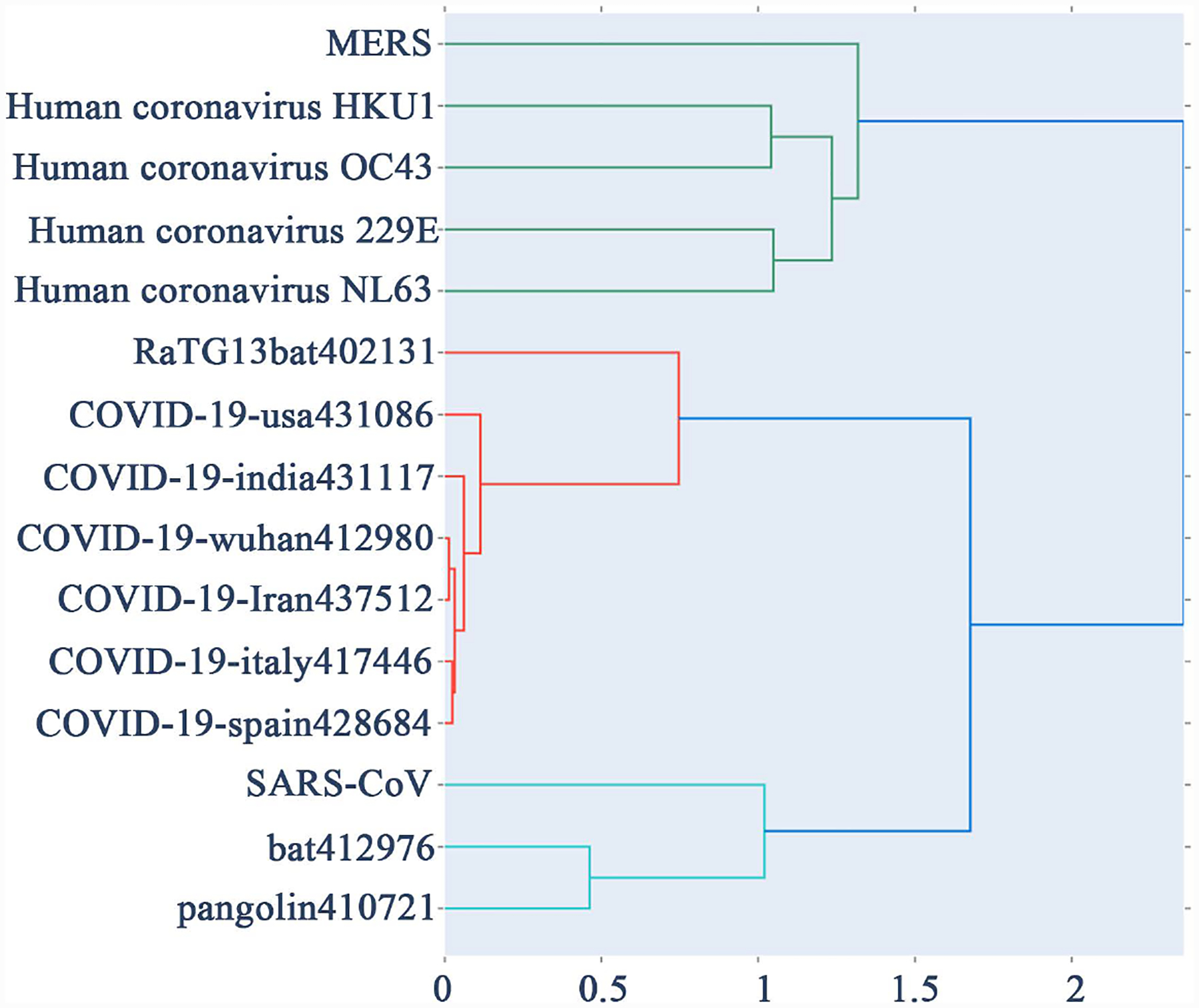
新型冠状病毒(SARS-COV-2)一般被称为 Covid-19 病毒,已传播到 213 个国家,确诊病例近 700 万例,死亡近 40 万人。此类重大疫情的爆发需要对病毒的基因组序列进行分类并确定其来源,以便进行规划、遏制和治疗。基于上述需求,我们报告了两种结合 CGR 进行聚类分析并在此基础上创建系统发生树的无比对方法。我们为每个 DNA 序列关联一个矩阵,然后将两个 DNA 序列之间的距离定义为其关联矩阵之间的距离。这些方法被用于冠状病毒序列的系统发生分析。我们的方法为分析和注释基因组及其系统发育关系提供了强大的工具。我们还将我们的工具与 ClustalX 算法进行了比较,后者是最流行的比对方法之一。结果表明,我们的无比对方法能够找到冠状病毒的近缘基因。
Similarity Studies of Corona Viruses through Chaos Game Representation.
The novel coronavirus (SARS-COV-2) is generally referred to as Covid-19 virus has spread to 213 countries with nearly 7 million confirmed cases and nearly 400,000 deaths. Such major outbreaks demand classification and origin of the virus genomic sequence, for planning, containment, and treatment. Motivated by the above need, we report two alignment-free methods combing with CGR to perform clustering analysis and create a phylogenetic tree based on it. To each DNA sequence we associate a matrix then define distance between two DNA sequences to be the distance between their associated matrix. These methods are being used for phylogenetic analysis of coronavirus sequences. Our approach provides a powerful tool for analyzing and annotating genomes and their phylogenetic relationships. We also compare our tool to ClustalX algorithm which is one of the most popular alignment methods. Our alignment-free methods are shown to be capable of finding closest genetic relatives of coronaviruses.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


