Cedric Landerer, Brian C O'Meara, Russell Zaretzki, Michael A Gilchrist
{"title":"利用突变选择模型解锁kluyveri Lachancea密码子渗入信号。","authors":"Cedric Landerer, Brian C O'Meara, Russell Zaretzki, Michael A Gilchrist","doi":"10.1186/s12862-020-01649-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>For decades, codon usage has been used as a measure of adaptation for translational efficiency and translation accuracy of a gene's coding sequence. These patterns of codon usage reflect both the selective and mutational environment in which the coding sequences evolved. Over this same period, gene transfer between lineages has become widely recognized as an important biological phenomenon. Nevertheless, most studies of codon usage implicitly assume that all genes within a genome evolved under the same selective and mutational environment, an assumption violated when introgression occurs. In order to better understand the effects of introgression on codon usage patterns and vice versa, we examine the patterns of codon usage in Lachancea kluyveri, a yeast which has experienced a large introgression. We quantify the effects of mutation bias and selection for translation efficiency on the codon usage pattern of the endogenous and introgressed exogenous genes using a Bayesian mixture model, ROC SEMPPR, which is built on mechanistic assumptions about protein synthesis and grounded in population genetics.</p><p><strong>Results: </strong>We find substantial differences in codon usage between the endogenous and exogenous genes, and show that these differences can be largely attributed to differences in mutation bias favoring A/T ending codons in the endogenous genes while favoring C/G ending codons in the exogenous genes. Recognizing the two different signatures of mutation bias and selection improves our ability to predict protein synthesis rate by 42% and allowed us to accurately assess the decaying signal of endogenous codon mutation and preferences. In addition, using our estimates of mutation bias and selection, we identify Eremothecium gossypii as the closest relative to the exogenous genes, providing an alternative hypothesis about the origin of the exogenous genes, estimate that the introgression occurred ∼6×10<sup>8</sup> generation ago, and estimate its historic and current selection against mismatched codon usage.</p><p><strong>Conclusions: </strong>Our work illustrates how mechanistic, population genetic models like ROC SEMPPR can separate the effects of mutation and selection on codon usage and provide quantitative estimates from sequence data.</p>","PeriodicalId":9111,"journal":{"name":"BMC Evolutionary Biology","volume":"20 1","pages":"109"},"PeriodicalIF":3.4000,"publicationDate":"2020-08-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12862-020-01649-w","citationCount":"3","resultStr":"{\"title\":\"Unlocking a signal of introgression from codons in Lachancea kluyveri using a mutation-selection model.\",\"authors\":\"Cedric Landerer, Brian C O'Meara, Russell Zaretzki, Michael A Gilchrist\",\"doi\":\"10.1186/s12862-020-01649-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>For decades, codon usage has been used as a measure of adaptation for translational efficiency and translation accuracy of a gene's coding sequence. These patterns of codon usage reflect both the selective and mutational environment in which the coding sequences evolved. Over this same period, gene transfer between lineages has become widely recognized as an important biological phenomenon. Nevertheless, most studies of codon usage implicitly assume that all genes within a genome evolved under the same selective and mutational environment, an assumption violated when introgression occurs. In order to better understand the effects of introgression on codon usage patterns and vice versa, we examine the patterns of codon usage in Lachancea kluyveri, a yeast which has experienced a large introgression. We quantify the effects of mutation bias and selection for translation efficiency on the codon usage pattern of the endogenous and introgressed exogenous genes using a Bayesian mixture model, ROC SEMPPR, which is built on mechanistic assumptions about protein synthesis and grounded in population genetics.</p><p><strong>Results: </strong>We find substantial differences in codon usage between the endogenous and exogenous genes, and show that these differences can be largely attributed to differences in mutation bias favoring A/T ending codons in the endogenous genes while favoring C/G ending codons in the exogenous genes. Recognizing the two different signatures of mutation bias and selection improves our ability to predict protein synthesis rate by 42% and allowed us to accurately assess the decaying signal of endogenous codon mutation and preferences. In addition, using our estimates of mutation bias and selection, we identify Eremothecium gossypii as the closest relative to the exogenous genes, providing an alternative hypothesis about the origin of the exogenous genes, estimate that the introgression occurred ∼6×10<sup>8</sup> generation ago, and estimate its historic and current selection against mismatched codon usage.</p><p><strong>Conclusions: </strong>Our work illustrates how mechanistic, population genetic models like ROC SEMPPR can separate the effects of mutation and selection on codon usage and provide quantitative estimates from sequence data.</p>\",\"PeriodicalId\":9111,\"journal\":{\"name\":\"BMC Evolutionary Biology\",\"volume\":\"20 1\",\"pages\":\"109\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2020-08-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12862-020-01649-w\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Evolutionary Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s12862-020-01649-w\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Evolutionary Biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12862-020-01649-w","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
Unlocking a signal of introgression from codons in Lachancea kluyveri using a mutation-selection model.
Background: For decades, codon usage has been used as a measure of adaptation for translational efficiency and translation accuracy of a gene's coding sequence. These patterns of codon usage reflect both the selective and mutational environment in which the coding sequences evolved. Over this same period, gene transfer between lineages has become widely recognized as an important biological phenomenon. Nevertheless, most studies of codon usage implicitly assume that all genes within a genome evolved under the same selective and mutational environment, an assumption violated when introgression occurs. In order to better understand the effects of introgression on codon usage patterns and vice versa, we examine the patterns of codon usage in Lachancea kluyveri, a yeast which has experienced a large introgression. We quantify the effects of mutation bias and selection for translation efficiency on the codon usage pattern of the endogenous and introgressed exogenous genes using a Bayesian mixture model, ROC SEMPPR, which is built on mechanistic assumptions about protein synthesis and grounded in population genetics.
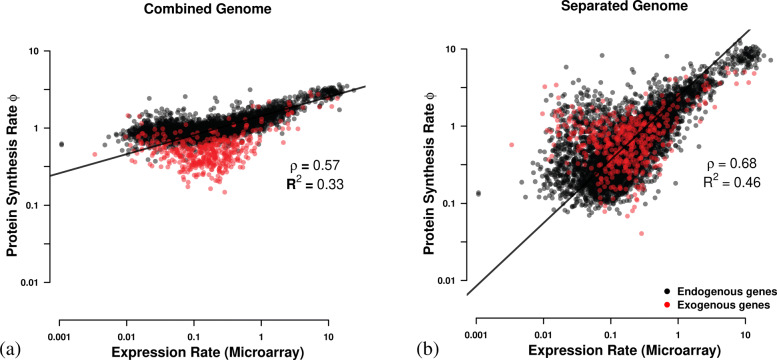
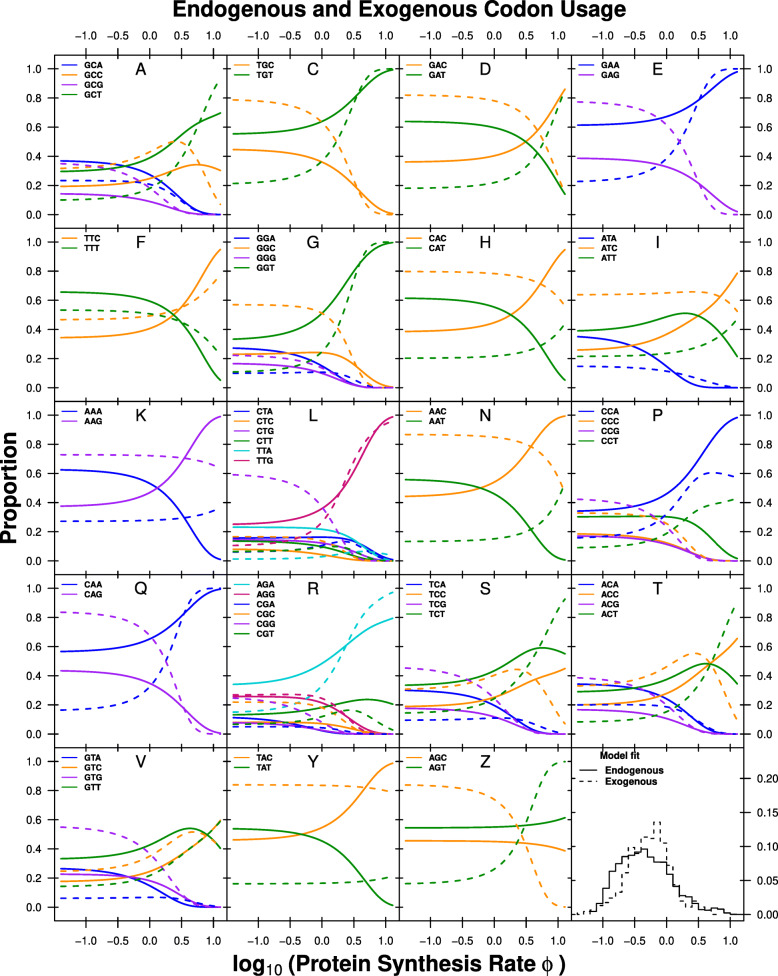
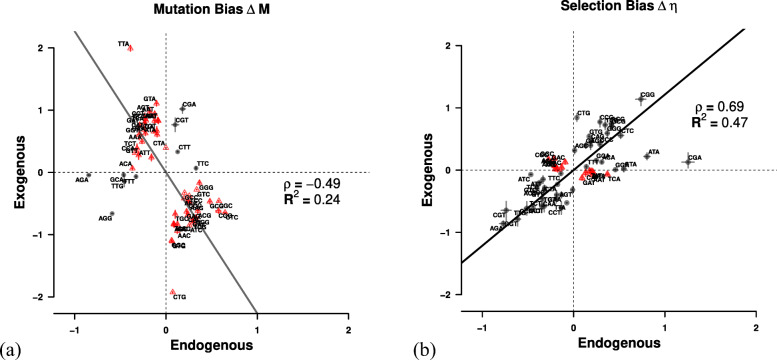
Results: We find substantial differences in codon usage between the endogenous and exogenous genes, and show that these differences can be largely attributed to differences in mutation bias favoring A/T ending codons in the endogenous genes while favoring C/G ending codons in the exogenous genes. Recognizing the two different signatures of mutation bias and selection improves our ability to predict protein synthesis rate by 42% and allowed us to accurately assess the decaying signal of endogenous codon mutation and preferences. In addition, using our estimates of mutation bias and selection, we identify Eremothecium gossypii as the closest relative to the exogenous genes, providing an alternative hypothesis about the origin of the exogenous genes, estimate that the introgression occurred ∼6×108 generation ago, and estimate its historic and current selection against mismatched codon usage.
Conclusions: Our work illustrates how mechanistic, population genetic models like ROC SEMPPR can separate the effects of mutation and selection on codon usage and provide quantitative estimates from sequence data.
期刊介绍:
BMC Evolutionary Biology is an open access, peer-reviewed journal that considers articles on all aspects of molecular and non-molecular evolution of all organisms, as well as phylogenetics and palaeontology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


