{"title":"超顺磁性氧化铁纳米颗粒促进缺血心肌细胞铁下垂。","authors":"Hao Zheng, Jieyun You, Xiaobo Yao, Qizheng Lu, Wei Guo, Yunli Shen","doi":"10.1111/jcmm.15722","DOIUrl":null,"url":null,"abstract":"Superparamagnetic iron oxide nanoparticles (SPION) have been widely used in the diagnosis and treatment for cardiovascular diseases.1-6 Correspondingly, the myocardial tissue safety of SPION is becoming a bottleneck to seriously restrict its clinical translation. In recent years, in vitro and in vivo experiments have confirmed that SPION-induced oxidative stress of normal myocardium in mice, leading to myocardial cell injury, apoptosis or necrosis.7-9 More alarmingly, SPION applied to ischemic myocardium could accumulate in the target sites for a long time with high concentration,5,6,10 thereby probably further aggravating oxidative stress injury and cardiomyocytes death.11,12 So far, however, the specific molecular mechanism of cardiotoxicity of SPION remains unclear. Previous studies have reported that SPION-induced apoptosis of murine macrophage (J774) cells 13 and necrosis of human endothelial cells.14 SPION can selectively induce autophagy-mediated cell death of human cancer cells (A549).15 After SPION pre-treatment, H9C2 cardiomyocytes were exposed to acrolein or H2O2, leading to reactive oxygen species (ROS) dependent cell necrosis.7 Our in vitro experiment showed that SPION significantly increased oxidative stress damage to overactivate autophagy and endoplasmic reticulum stress, eventually resulting in cardiomyocyte apoptosis.12 Furthermore, SPION could elicit IL-1βrelease and pyroptosis in macrophages, especially with the octapod and plate morphology.16 Notably, it has been recently reported that sorafenib or cisplatin assembled into nano-devices containing SPION, which are phagocytized by tumour cells and degraded into free divalent iron to accelerate Fenton reaction, leading to the lipid peroxidation burst to promote ferroptosis of tumour cells.17,18 Taken together, SPION can induce apoptosis, necrosis, autophagy, pyroptosis or ferroptosis in vitro and in vivo studies. The discrepancy may be attributed to distinct cell types and experiments design. It has already been well documented that the toxicity of SPION is mainly due to its degradation and release of free iron to catalyse Fenton reaction, leading to oxidative stress by a large number of ROS generation.19,20 Then, what is the downstream molecular mechanism of SPION mediated cardiotoxicity? Ferroptosis is a novel form of regulated cell death characterized by the iron-dependent accumulation of lipid peroxides to lethal levels, which is morphologically, biochemically, and genetically distinct from apoptosis, necroptosis and autophagy.21 Recent studies found that ferroptosis is not only an important pathological mechanism in the case of circulating iron overload of hemochromatosis,22 but also a key molecular mechanism of cellular iron overload in doxorubicin (DOX) induced cardiomyopathy.23 DOX induced mitochondria iron overload by down-regulating ABCB8,24 a mitochondrial protein that facilitates iron export, to elicit lipid peroxidation and mitochondria dysfunction, eventually causing cardiomyocytes ferroptosis.23 Mice that were subjected to 30 minutes of myocardial ischemia followed by 24 hours of reperfusion had significantly higher levels of cardiac non-heme iron, cardiac ferritin H, ferritin L and Ptgs2 mRNA. Both ferroptosis inhibitor Ferrostatin-1 (Fer-1) and iron chelator Dexrazoxane (DXZ) pre-treatment significantly reduced I/R-induced cardiac remodelling and fibrosis, indicating that ischemia-reperfusion could also induce cardiomyocytes iron overload to cause ferroptosis and subsequent left ventricular remodelling.23 Myocardial haemorrhage is a frequent complication after successful myocardial reperfusion,25,26 which is associated with residual myocardial iron in post-myocardial infarction (MI) patients received reperfusion therapy.27 It is reasonable to infer that this iron accumulation has a potential to generate excessive ROS and trigger pathological events such as ferroptosis. A previous study also confirmed that ferroptosis is a significant type of cell death in cardiomyocytes; moreover, mechanistic target of rapamycin (mTOR) was found to play an important role in protecting cardiomyocytes against excess iron and ferroptosis by regulating ROS production.28 In addition, glutathione peroxidase 4 (GPX4), which protects cells from ferroptosis, was down-regulated in the early and middle stages of MI mouse model, suggesting that ferroptosis during MI was in part due to a reduction in GPX4 protein.29 Even though signalling pathways of ferroptosis in cardiovascular diseases is not yet well characterized, it has been confirmed that ischemia-reperfusion (I/R) could induce mitochondrial iron overload in cardiomyocytes rather than the increase of iron content in cytoplasm.30 In this study, mice treated with 2,2′-bipyridyl (BPD), which has high membrane permeability and thus is able to access","PeriodicalId":15215,"journal":{"name":"Journal of Cellular and Molecular Medicine","volume":null,"pages":null},"PeriodicalIF":5.3000,"publicationDate":"2020-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1111/jcmm.15722","citationCount":"17","resultStr":"{\"title\":\"Superparamagnetic iron oxide nanoparticles promote ferroptosis of ischemic cardiomyocytes.\",\"authors\":\"Hao Zheng, Jieyun You, Xiaobo Yao, Qizheng Lu, Wei Guo, Yunli Shen\",\"doi\":\"10.1111/jcmm.15722\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Superparamagnetic iron oxide nanoparticles (SPION) have been widely used in the diagnosis and treatment for cardiovascular diseases.1-6 Correspondingly, the myocardial tissue safety of SPION is becoming a bottleneck to seriously restrict its clinical translation. In recent years, in vitro and in vivo experiments have confirmed that SPION-induced oxidative stress of normal myocardium in mice, leading to myocardial cell injury, apoptosis or necrosis.7-9 More alarmingly, SPION applied to ischemic myocardium could accumulate in the target sites for a long time with high concentration,5,6,10 thereby probably further aggravating oxidative stress injury and cardiomyocytes death.11,12 So far, however, the specific molecular mechanism of cardiotoxicity of SPION remains unclear. Previous studies have reported that SPION-induced apoptosis of murine macrophage (J774) cells 13 and necrosis of human endothelial cells.14 SPION can selectively induce autophagy-mediated cell death of human cancer cells (A549).15 After SPION pre-treatment, H9C2 cardiomyocytes were exposed to acrolein or H2O2, leading to reactive oxygen species (ROS) dependent cell necrosis.7 Our in vitro experiment showed that SPION significantly increased oxidative stress damage to overactivate autophagy and endoplasmic reticulum stress, eventually resulting in cardiomyocyte apoptosis.12 Furthermore, SPION could elicit IL-1βrelease and pyroptosis in macrophages, especially with the octapod and plate morphology.16 Notably, it has been recently reported that sorafenib or cisplatin assembled into nano-devices containing SPION, which are phagocytized by tumour cells and degraded into free divalent iron to accelerate Fenton reaction, leading to the lipid peroxidation burst to promote ferroptosis of tumour cells.17,18 Taken together, SPION can induce apoptosis, necrosis, autophagy, pyroptosis or ferroptosis in vitro and in vivo studies. The discrepancy may be attributed to distinct cell types and experiments design. It has already been well documented that the toxicity of SPION is mainly due to its degradation and release of free iron to catalyse Fenton reaction, leading to oxidative stress by a large number of ROS generation.19,20 Then, what is the downstream molecular mechanism of SPION mediated cardiotoxicity? Ferroptosis is a novel form of regulated cell death characterized by the iron-dependent accumulation of lipid peroxides to lethal levels, which is morphologically, biochemically, and genetically distinct from apoptosis, necroptosis and autophagy.21 Recent studies found that ferroptosis is not only an important pathological mechanism in the case of circulating iron overload of hemochromatosis,22 but also a key molecular mechanism of cellular iron overload in doxorubicin (DOX) induced cardiomyopathy.23 DOX induced mitochondria iron overload by down-regulating ABCB8,24 a mitochondrial protein that facilitates iron export, to elicit lipid peroxidation and mitochondria dysfunction, eventually causing cardiomyocytes ferroptosis.23 Mice that were subjected to 30 minutes of myocardial ischemia followed by 24 hours of reperfusion had significantly higher levels of cardiac non-heme iron, cardiac ferritin H, ferritin L and Ptgs2 mRNA. Both ferroptosis inhibitor Ferrostatin-1 (Fer-1) and iron chelator Dexrazoxane (DXZ) pre-treatment significantly reduced I/R-induced cardiac remodelling and fibrosis, indicating that ischemia-reperfusion could also induce cardiomyocytes iron overload to cause ferroptosis and subsequent left ventricular remodelling.23 Myocardial haemorrhage is a frequent complication after successful myocardial reperfusion,25,26 which is associated with residual myocardial iron in post-myocardial infarction (MI) patients received reperfusion therapy.27 It is reasonable to infer that this iron accumulation has a potential to generate excessive ROS and trigger pathological events such as ferroptosis. A previous study also confirmed that ferroptosis is a significant type of cell death in cardiomyocytes; moreover, mechanistic target of rapamycin (mTOR) was found to play an important role in protecting cardiomyocytes against excess iron and ferroptosis by regulating ROS production.28 In addition, glutathione peroxidase 4 (GPX4), which protects cells from ferroptosis, was down-regulated in the early and middle stages of MI mouse model, suggesting that ferroptosis during MI was in part due to a reduction in GPX4 protein.29 Even though signalling pathways of ferroptosis in cardiovascular diseases is not yet well characterized, it has been confirmed that ischemia-reperfusion (I/R) could induce mitochondrial iron overload in cardiomyocytes rather than the increase of iron content in cytoplasm.30 In this study, mice treated with 2,2′-bipyridyl (BPD), which has high membrane permeability and thus is able to access\",\"PeriodicalId\":15215,\"journal\":{\"name\":\"Journal of Cellular and Molecular Medicine\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2020-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1111/jcmm.15722\",\"citationCount\":\"17\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Cellular and Molecular Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1111/jcmm.15722\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2020/8/11 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Cellular and Molecular Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1111/jcmm.15722","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/8/11 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Superparamagnetic iron oxide nanoparticles promote ferroptosis of ischemic cardiomyocytes.
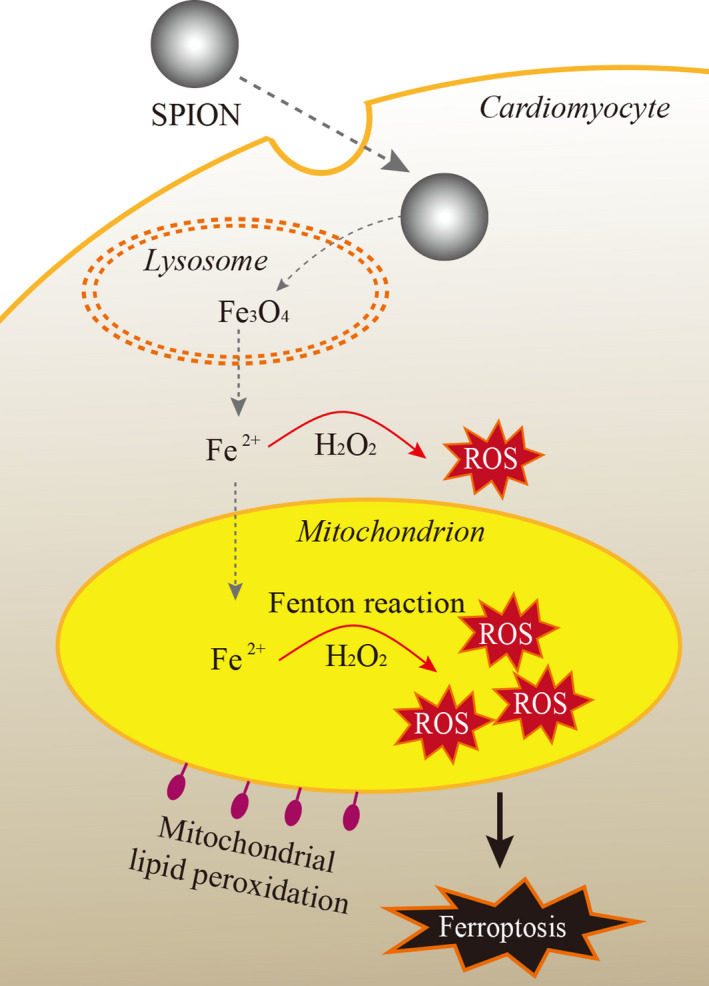
Superparamagnetic iron oxide nanoparticles (SPION) have been widely used in the diagnosis and treatment for cardiovascular diseases.1-6 Correspondingly, the myocardial tissue safety of SPION is becoming a bottleneck to seriously restrict its clinical translation. In recent years, in vitro and in vivo experiments have confirmed that SPION-induced oxidative stress of normal myocardium in mice, leading to myocardial cell injury, apoptosis or necrosis.7-9 More alarmingly, SPION applied to ischemic myocardium could accumulate in the target sites for a long time with high concentration,5,6,10 thereby probably further aggravating oxidative stress injury and cardiomyocytes death.11,12 So far, however, the specific molecular mechanism of cardiotoxicity of SPION remains unclear. Previous studies have reported that SPION-induced apoptosis of murine macrophage (J774) cells 13 and necrosis of human endothelial cells.14 SPION can selectively induce autophagy-mediated cell death of human cancer cells (A549).15 After SPION pre-treatment, H9C2 cardiomyocytes were exposed to acrolein or H2O2, leading to reactive oxygen species (ROS) dependent cell necrosis.7 Our in vitro experiment showed that SPION significantly increased oxidative stress damage to overactivate autophagy and endoplasmic reticulum stress, eventually resulting in cardiomyocyte apoptosis.12 Furthermore, SPION could elicit IL-1βrelease and pyroptosis in macrophages, especially with the octapod and plate morphology.16 Notably, it has been recently reported that sorafenib or cisplatin assembled into nano-devices containing SPION, which are phagocytized by tumour cells and degraded into free divalent iron to accelerate Fenton reaction, leading to the lipid peroxidation burst to promote ferroptosis of tumour cells.17,18 Taken together, SPION can induce apoptosis, necrosis, autophagy, pyroptosis or ferroptosis in vitro and in vivo studies. The discrepancy may be attributed to distinct cell types and experiments design. It has already been well documented that the toxicity of SPION is mainly due to its degradation and release of free iron to catalyse Fenton reaction, leading to oxidative stress by a large number of ROS generation.19,20 Then, what is the downstream molecular mechanism of SPION mediated cardiotoxicity? Ferroptosis is a novel form of regulated cell death characterized by the iron-dependent accumulation of lipid peroxides to lethal levels, which is morphologically, biochemically, and genetically distinct from apoptosis, necroptosis and autophagy.21 Recent studies found that ferroptosis is not only an important pathological mechanism in the case of circulating iron overload of hemochromatosis,22 but also a key molecular mechanism of cellular iron overload in doxorubicin (DOX) induced cardiomyopathy.23 DOX induced mitochondria iron overload by down-regulating ABCB8,24 a mitochondrial protein that facilitates iron export, to elicit lipid peroxidation and mitochondria dysfunction, eventually causing cardiomyocytes ferroptosis.23 Mice that were subjected to 30 minutes of myocardial ischemia followed by 24 hours of reperfusion had significantly higher levels of cardiac non-heme iron, cardiac ferritin H, ferritin L and Ptgs2 mRNA. Both ferroptosis inhibitor Ferrostatin-1 (Fer-1) and iron chelator Dexrazoxane (DXZ) pre-treatment significantly reduced I/R-induced cardiac remodelling and fibrosis, indicating that ischemia-reperfusion could also induce cardiomyocytes iron overload to cause ferroptosis and subsequent left ventricular remodelling.23 Myocardial haemorrhage is a frequent complication after successful myocardial reperfusion,25,26 which is associated with residual myocardial iron in post-myocardial infarction (MI) patients received reperfusion therapy.27 It is reasonable to infer that this iron accumulation has a potential to generate excessive ROS and trigger pathological events such as ferroptosis. A previous study also confirmed that ferroptosis is a significant type of cell death in cardiomyocytes; moreover, mechanistic target of rapamycin (mTOR) was found to play an important role in protecting cardiomyocytes against excess iron and ferroptosis by regulating ROS production.28 In addition, glutathione peroxidase 4 (GPX4), which protects cells from ferroptosis, was down-regulated in the early and middle stages of MI mouse model, suggesting that ferroptosis during MI was in part due to a reduction in GPX4 protein.29 Even though signalling pathways of ferroptosis in cardiovascular diseases is not yet well characterized, it has been confirmed that ischemia-reperfusion (I/R) could induce mitochondrial iron overload in cardiomyocytes rather than the increase of iron content in cytoplasm.30 In this study, mice treated with 2,2′-bipyridyl (BPD), which has high membrane permeability and thus is able to access
期刊介绍:
Bridging physiology and cellular medicine, and molecular biology and molecular therapeutics, Journal of Cellular and Molecular Medicine publishes basic research that furthers our understanding of the cellular and molecular mechanisms of disease and translational studies that convert this knowledge into therapeutic approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


