Yingjie Zhou, Muhammad Tariq, Sijie He, Uzma Abdullah, Jianguo Zhang, Shahid Mahmood Baig
{"title":"全外显子组测序确定了导致五个巴基斯坦近亲家庭听力损失的突变。","authors":"Yingjie Zhou, Muhammad Tariq, Sijie He, Uzma Abdullah, Jianguo Zhang, Shahid Mahmood Baig","doi":"10.1186/s12881-020-01087-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Hearing loss is the most common sensory defect, and it affects over 6% of the population worldwide. Approximately 50-60% of hearing loss patients are attributed to genetic causes. Currently, more than 100 genes have been reported to cause non-syndromic hearing loss. It is possible and efficient to screen all potential disease-causing genes for hereditary hearing loss by whole exome sequencing (WES).</p><p><strong>Methods: </strong>We collected 5 consanguineous pedigrees from Pakistan with hearing loss and applied WES in selected patients for each pedigree, followed by bioinformatics analysis and Sanger validation to identify the causal genes.</p><p><strong>Results: </strong>Variants in 7 genes were identified and validated in these pedigrees. We identified single candidate variant for 3 pedigrees: GIPC3 (c.937 T > C), LOXHD1 (c.6136G > A) and TMPRSS3 (c.941 T > C). The remaining 2 pedigrees each contained two candidate variants: TECTA (c.4045G > A) and MYO15A (c.3310G > T and c.9913G > C) for one pedigree and DFNB59 (c.494G > A) and TRIOBP (c.1952C > T) for the other pedigree. The candidate variants were validated in all available samples by Sanger sequencing.</p><p><strong>Conclusion: </strong>The candidate variants in hearing-loss genes were validated to be co-segregated in the pedigrees, and they may indicate the aetiologies of hearing loss in such patients. We also suggest that WES may be a suitable strategy for hearing-loss gene screening in clinical detection.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"151"},"PeriodicalIF":0.0000,"publicationDate":"2020-07-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01087-x","citationCount":"8","resultStr":"{\"title\":\"Whole exome sequencing identified mutations causing hearing loss in five consanguineous Pakistani families.\",\"authors\":\"Yingjie Zhou, Muhammad Tariq, Sijie He, Uzma Abdullah, Jianguo Zhang, Shahid Mahmood Baig\",\"doi\":\"10.1186/s12881-020-01087-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Hearing loss is the most common sensory defect, and it affects over 6% of the population worldwide. Approximately 50-60% of hearing loss patients are attributed to genetic causes. Currently, more than 100 genes have been reported to cause non-syndromic hearing loss. It is possible and efficient to screen all potential disease-causing genes for hereditary hearing loss by whole exome sequencing (WES).</p><p><strong>Methods: </strong>We collected 5 consanguineous pedigrees from Pakistan with hearing loss and applied WES in selected patients for each pedigree, followed by bioinformatics analysis and Sanger validation to identify the causal genes.</p><p><strong>Results: </strong>Variants in 7 genes were identified and validated in these pedigrees. We identified single candidate variant for 3 pedigrees: GIPC3 (c.937 T > C), LOXHD1 (c.6136G > A) and TMPRSS3 (c.941 T > C). The remaining 2 pedigrees each contained two candidate variants: TECTA (c.4045G > A) and MYO15A (c.3310G > T and c.9913G > C) for one pedigree and DFNB59 (c.494G > A) and TRIOBP (c.1952C > T) for the other pedigree. The candidate variants were validated in all available samples by Sanger sequencing.</p><p><strong>Conclusion: </strong>The candidate variants in hearing-loss genes were validated to be co-segregated in the pedigrees, and they may indicate the aetiologies of hearing loss in such patients. We also suggest that WES may be a suitable strategy for hearing-loss gene screening in clinical detection.</p>\",\"PeriodicalId\":9015,\"journal\":{\"name\":\"BMC Medical Genetics\",\"volume\":\" \",\"pages\":\"151\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-07-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12881-020-01087-x\",\"citationCount\":\"8\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12881-020-01087-x\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01087-x","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
Whole exome sequencing identified mutations causing hearing loss in five consanguineous Pakistani families.
Background: Hearing loss is the most common sensory defect, and it affects over 6% of the population worldwide. Approximately 50-60% of hearing loss patients are attributed to genetic causes. Currently, more than 100 genes have been reported to cause non-syndromic hearing loss. It is possible and efficient to screen all potential disease-causing genes for hereditary hearing loss by whole exome sequencing (WES).
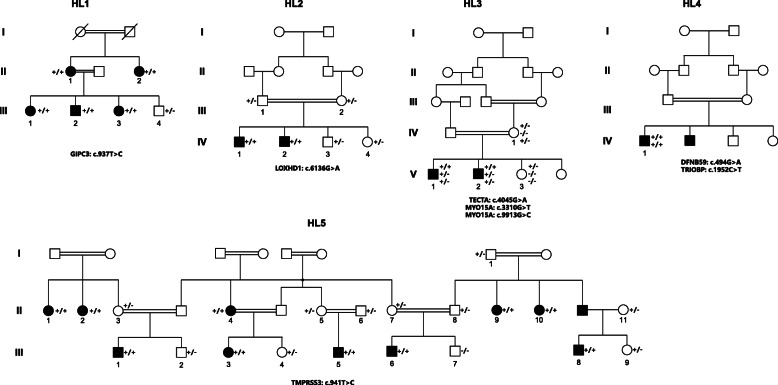
Methods: We collected 5 consanguineous pedigrees from Pakistan with hearing loss and applied WES in selected patients for each pedigree, followed by bioinformatics analysis and Sanger validation to identify the causal genes.
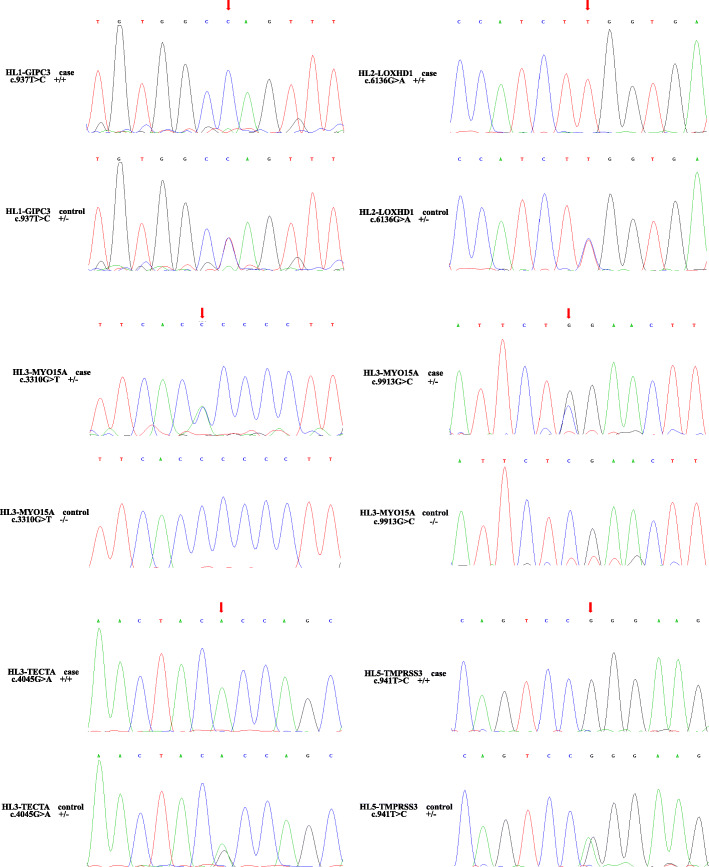
Results: Variants in 7 genes were identified and validated in these pedigrees. We identified single candidate variant for 3 pedigrees: GIPC3 (c.937 T > C), LOXHD1 (c.6136G > A) and TMPRSS3 (c.941 T > C). The remaining 2 pedigrees each contained two candidate variants: TECTA (c.4045G > A) and MYO15A (c.3310G > T and c.9913G > C) for one pedigree and DFNB59 (c.494G > A) and TRIOBP (c.1952C > T) for the other pedigree. The candidate variants were validated in all available samples by Sanger sequencing.
Conclusion: The candidate variants in hearing-loss genes were validated to be co-segregated in the pedigrees, and they may indicate the aetiologies of hearing loss in such patients. We also suggest that WES may be a suitable strategy for hearing-loss gene screening in clinical detection.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


