Dörte Wittenburg, Sarah Bonk, Michael Doschoris, Henry Reyer
{"title":"牲畜种群数量性状位点精细图谱的实验设计。","authors":"Dörte Wittenburg, Sarah Bonk, Michael Doschoris, Henry Reyer","doi":"10.1186/s12863-020-00871-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Single nucleotide polymorphisms (SNPs) which capture a significant impact on a trait can be identified with genome-wide association studies. High linkage disequilibrium (LD) among SNPs makes it difficult to identify causative variants correctly. Thus, often target regions instead of single SNPs are reported. Sample size has not only a crucial impact on the precision of parameter estimates, it also ensures that a desired level of statistical power can be reached. We study the design of experiments for fine-mapping of signals of a quantitative trait locus in such a target region.</p><p><strong>Methods: </strong>A multi-locus model allows to identify causative variants simultaneously, to state their positions more precisely and to account for existing dependencies. Based on the commonly applied SNP-BLUP approach, we determine the z-score statistic for locally testing non-zero SNP effects and investigate its distribution under the alternative hypothesis. This quantity employs the theoretical instead of observed dependence between SNPs; it can be set up as a function of paternal and maternal LD for any given population structure.</p><p><strong>Results: </strong>We simulated multiple paternal half-sib families and considered a target region of 1 Mbp. A bimodal distribution of estimated sample size was observed, particularly if more than two causative variants were assumed. The median of estimates constituted the final proposal of optimal sample size; it was consistently less than sample size estimated from single-SNP investigation which was used as a baseline approach. The second mode pointed to inflated sample sizes and could be explained by blocks of varying linkage phases leading to negative correlations between SNPs. Optimal sample size increased almost linearly with number of signals to be identified but depended much stronger on the assumption on heritability. For instance, three times as many samples were required if heritability was 0.1 compared to 0.3. An R package is provided that comprises all required tools.</p><p><strong>Conclusions: </strong>Our approach incorporates information about the population structure into the design of experiments. Compared to a conventional method, this leads to a reduced estimate of sample size enabling the resource-saving design of future experiments for fine-mapping of candidate variants.</p>","PeriodicalId":9197,"journal":{"name":"BMC Genetics","volume":" ","pages":"66"},"PeriodicalIF":2.9000,"publicationDate":"2020-06-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7324978/pdf/","citationCount":"0","resultStr":"{\"title\":\"Design of experiments for fine-mapping quantitative trait loci in livestock populations.\",\"authors\":\"Dörte Wittenburg, Sarah Bonk, Michael Doschoris, Henry Reyer\",\"doi\":\"10.1186/s12863-020-00871-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Single nucleotide polymorphisms (SNPs) which capture a significant impact on a trait can be identified with genome-wide association studies. High linkage disequilibrium (LD) among SNPs makes it difficult to identify causative variants correctly. Thus, often target regions instead of single SNPs are reported. Sample size has not only a crucial impact on the precision of parameter estimates, it also ensures that a desired level of statistical power can be reached. We study the design of experiments for fine-mapping of signals of a quantitative trait locus in such a target region.</p><p><strong>Methods: </strong>A multi-locus model allows to identify causative variants simultaneously, to state their positions more precisely and to account for existing dependencies. Based on the commonly applied SNP-BLUP approach, we determine the z-score statistic for locally testing non-zero SNP effects and investigate its distribution under the alternative hypothesis. This quantity employs the theoretical instead of observed dependence between SNPs; it can be set up as a function of paternal and maternal LD for any given population structure.</p><p><strong>Results: </strong>We simulated multiple paternal half-sib families and considered a target region of 1 Mbp. A bimodal distribution of estimated sample size was observed, particularly if more than two causative variants were assumed. The median of estimates constituted the final proposal of optimal sample size; it was consistently less than sample size estimated from single-SNP investigation which was used as a baseline approach. The second mode pointed to inflated sample sizes and could be explained by blocks of varying linkage phases leading to negative correlations between SNPs. Optimal sample size increased almost linearly with number of signals to be identified but depended much stronger on the assumption on heritability. For instance, three times as many samples were required if heritability was 0.1 compared to 0.3. An R package is provided that comprises all required tools.</p><p><strong>Conclusions: </strong>Our approach incorporates information about the population structure into the design of experiments. Compared to a conventional method, this leads to a reduced estimate of sample size enabling the resource-saving design of future experiments for fine-mapping of candidate variants.</p>\",\"PeriodicalId\":9197,\"journal\":{\"name\":\"BMC Genetics\",\"volume\":\" \",\"pages\":\"66\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2020-06-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7324978/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s12863-020-00871-1\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12863-020-00871-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
摘要
背景:单核苷酸多态性(SNPs)可通过全基因组关联研究识别出对性状有显著影响的变异。SNP 之间的高连锁不平衡(LD)使得正确识别致病变异非常困难。因此,通常报告的是目标区域而不是单个 SNP。样本量不仅对参数估计的精确度有重要影响,还能确保达到理想的统计能力水平。我们研究了在这种目标区域中精细绘制数量性状基因座信号的实验设计:方法:多焦点模型可以同时识别致病变异,更精确地确定其位置,并考虑现有的依赖关系。基于常用的 SNP-BLUP 方法,我们确定了用于局部检验非零 SNP 效应的 z 分统计量,并研究了其在替代假设下的分布情况。该统计量采用的是 SNP 之间的理论依赖关系,而不是观察到的依赖关系;它可以在任何给定的种群结构中设置为父系和母系 LD 的函数:我们模拟了多个父系半兄弟家族,并考虑了 1 Mbp 的目标区域。我们观察到估计样本大小呈双峰分布,尤其是假设有两个以上的致病变异时。估计值的中位数构成了最佳样本量的最终建议;它始终小于作为基线方法的单 SNP 调查估计的样本量。第二种模式表明样本量被夸大,其原因可能是不同连接阶段的区块导致 SNP 之间的负相关。最佳样本量与待鉴定信号的数量几乎呈线性增长,但对遗传性假设的依赖性更大。例如,如果遗传率为 0.1,所需的样本量是 0.3 的三倍。我们提供了一个包含所有必要工具的 R 软件包:我们的方法将种群结构信息纳入实验设计。与传统方法相比,这种方法减少了样本量的估算,从而可以节省资源,设计未来的候选变异体精细图谱实验。
Design of experiments for fine-mapping quantitative trait loci in livestock populations.
Background: Single nucleotide polymorphisms (SNPs) which capture a significant impact on a trait can be identified with genome-wide association studies. High linkage disequilibrium (LD) among SNPs makes it difficult to identify causative variants correctly. Thus, often target regions instead of single SNPs are reported. Sample size has not only a crucial impact on the precision of parameter estimates, it also ensures that a desired level of statistical power can be reached. We study the design of experiments for fine-mapping of signals of a quantitative trait locus in such a target region.
Methods: A multi-locus model allows to identify causative variants simultaneously, to state their positions more precisely and to account for existing dependencies. Based on the commonly applied SNP-BLUP approach, we determine the z-score statistic for locally testing non-zero SNP effects and investigate its distribution under the alternative hypothesis. This quantity employs the theoretical instead of observed dependence between SNPs; it can be set up as a function of paternal and maternal LD for any given population structure.
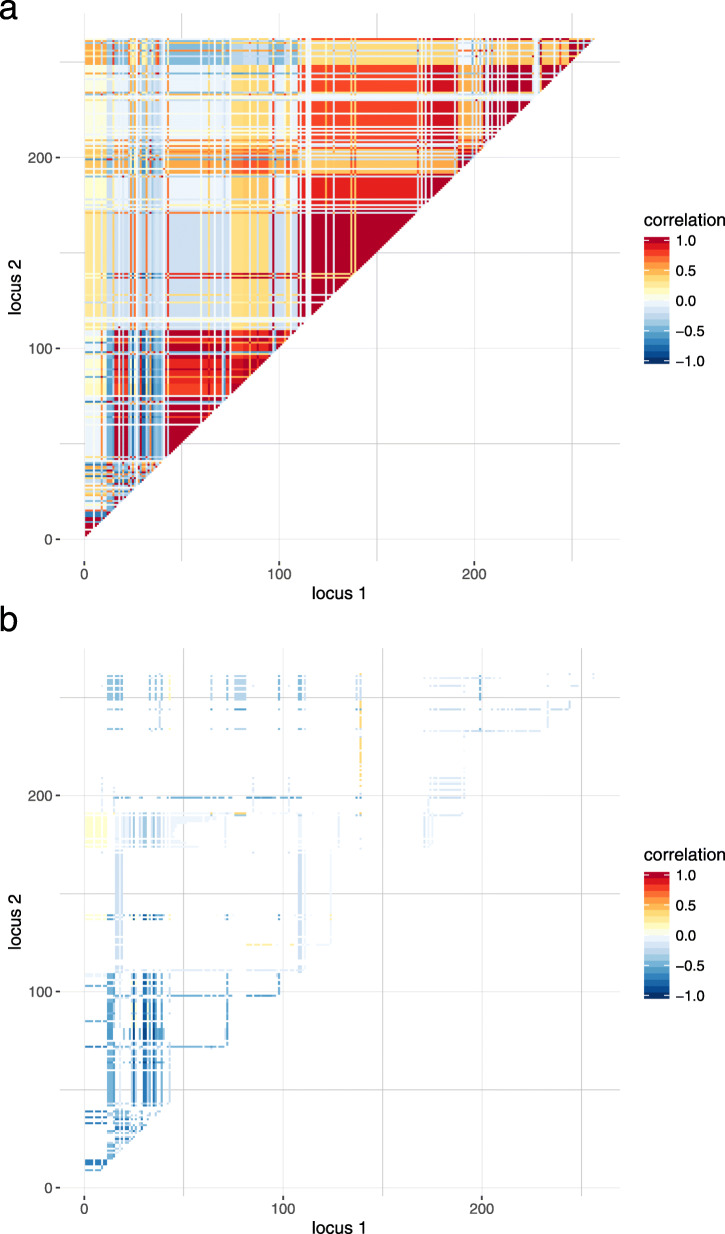
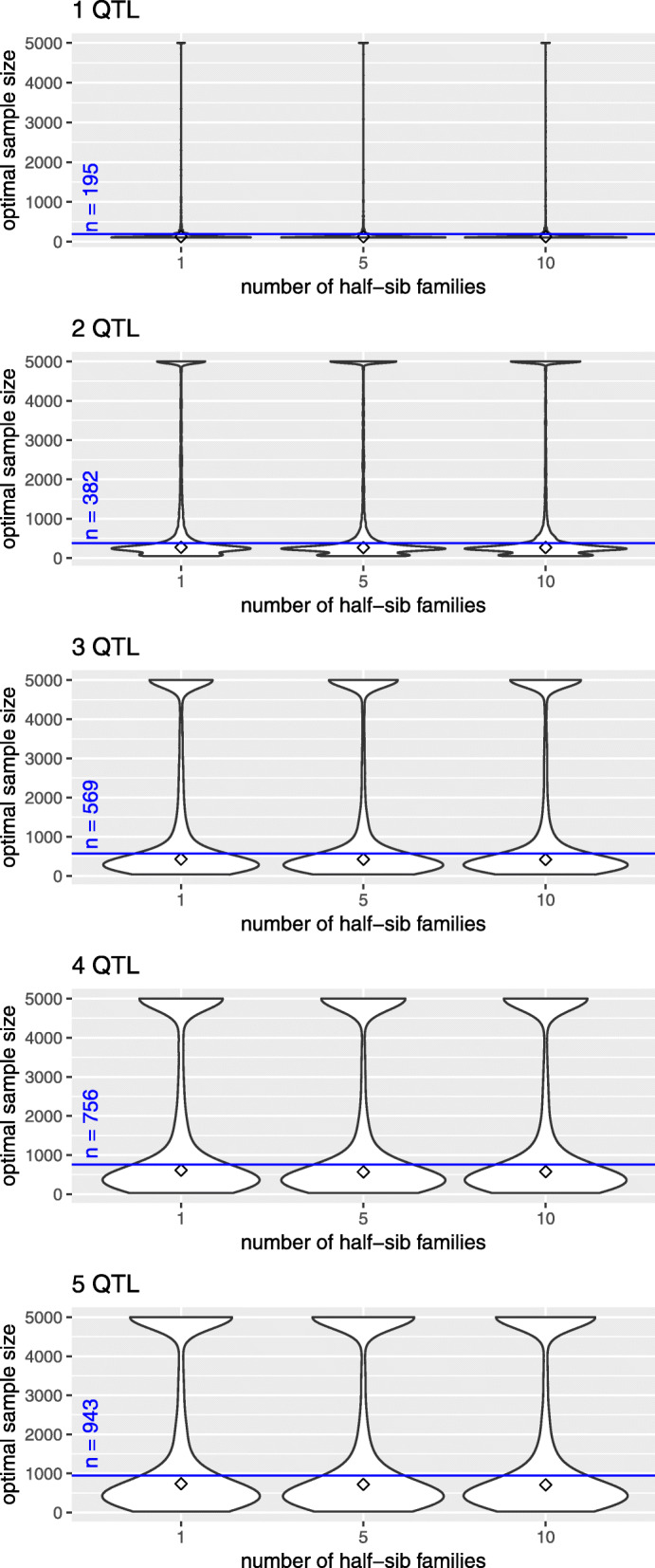
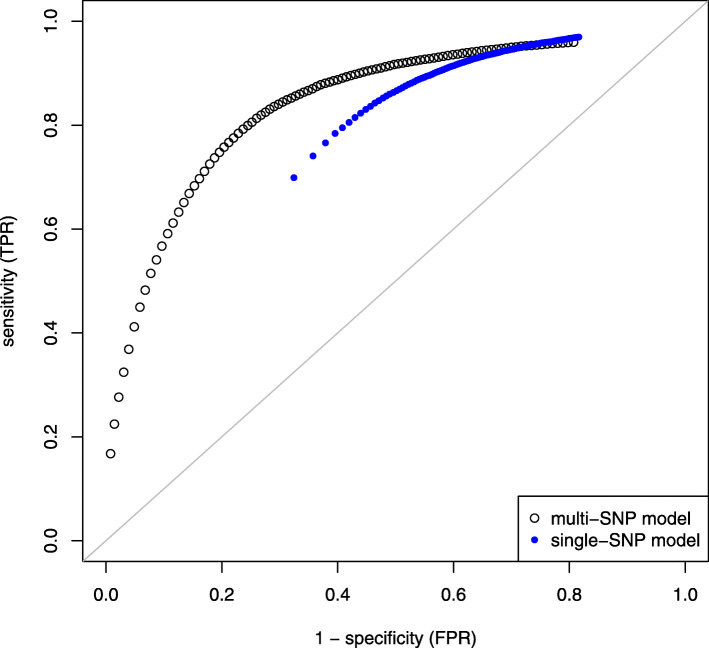
Results: We simulated multiple paternal half-sib families and considered a target region of 1 Mbp. A bimodal distribution of estimated sample size was observed, particularly if more than two causative variants were assumed. The median of estimates constituted the final proposal of optimal sample size; it was consistently less than sample size estimated from single-SNP investigation which was used as a baseline approach. The second mode pointed to inflated sample sizes and could be explained by blocks of varying linkage phases leading to negative correlations between SNPs. Optimal sample size increased almost linearly with number of signals to be identified but depended much stronger on the assumption on heritability. For instance, three times as many samples were required if heritability was 0.1 compared to 0.3. An R package is provided that comprises all required tools.
Conclusions: Our approach incorporates information about the population structure into the design of experiments. Compared to a conventional method, this leads to a reduced estimate of sample size enabling the resource-saving design of future experiments for fine-mapping of candidate variants.
期刊介绍:
BMC Genetics is an open access, peer-reviewed journal that considers articles on all aspects of inheritance and variation in individuals and among populations.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


