Valeska Stonawski, Jakob Roetner, Tamme W Goecke, Peter A Fasching, Matthias W Beckmann, Johannes Kornhuber, Oliver Kratz, Gunther H Moll, Anna Eichler, Hartmut Heinrich, Stefan Frey
{"title":"接受非药物治疗的产前抑郁症儿童的全基因组 DNA 甲基化模式:两个独立队列的研究结果","authors":"Valeska Stonawski, Jakob Roetner, Tamme W Goecke, Peter A Fasching, Matthias W Beckmann, Johannes Kornhuber, Oliver Kratz, Gunther H Moll, Anna Eichler, Hartmut Heinrich, Stefan Frey","doi":"10.1177/2516865720932146","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Maternal depressive symptoms are a common phenomenon during pregnancy and are related to negative outcomes for child development and health. Modifications in child DNA methylation are discussed as an underlying mechanism for the association between prenatal depressive symptoms and alterations in child outcomes. However, formerly reported genome-wide associations have yet to be replicated.</p><p><strong>Methods: </strong>In an epigenome-wide association study (EWAS), alterations of DNA methylation related to maternal prenatal depressive symptoms were investigated in buccal cell samples from 174 children (n = 52 exposed to prenatal depressive symptoms; 6-9 years old) of the German longitudinal study FRAMES-FRANCES. Whole blood samples from the independent, age-comparable ARIES subsample of the ARIES/ALSPAC study (n = 641; n = 159 exposed to prenatal depressive symptoms; 7-8 years old) were examined as a confirmation sample. Depressive symptoms were assessed with the Edinburgh Postnatal Depression Scale. DNA methylation was analyzed with the Infinium Human Methylation 450k BeadChip. Modifications in single CpGs, regions, and biological pathways were investigated. Results were adjusted for age and birth outcomes as well as postnatal and current maternal depressive symptoms. Analyses were performed for the whole sample as well as separated for sex.</p><p><strong>Results: </strong>The EWAS yielded no differentially methylated CpG or region as well as no accordance between samples withstanding correction for multiple testing. In pathway analyses, no overlapping functional domain was found to be enriched for either sample. A comparison of current and former findings suggests some overlapping methylation modifications from infancy to childhood. Results suggest that there might be sex-specific differential methylation, which should be further investigated in additional studies.</p><p><strong>Conclusions: </strong>The current, mainly nonsignificant, results challenge the assumption of consistent modifications of DNA methylation in children exposed to prenatal depressive symptoms. Despite the relatively small sample size used in this study, this lack of significant results may reflect diverse issues of environmental epigenetic studies, which need to be addressed in future research.</p>","PeriodicalId":41996,"journal":{"name":"Epigenetics Insights","volume":null,"pages":null},"PeriodicalIF":3.2000,"publicationDate":"2020-06-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/e5/0c/10.1177_2516865720932146.PMC7298426.pdf","citationCount":"0","resultStr":"{\"title\":\"Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts.\",\"authors\":\"Valeska Stonawski, Jakob Roetner, Tamme W Goecke, Peter A Fasching, Matthias W Beckmann, Johannes Kornhuber, Oliver Kratz, Gunther H Moll, Anna Eichler, Hartmut Heinrich, Stefan Frey\",\"doi\":\"10.1177/2516865720932146\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Maternal depressive symptoms are a common phenomenon during pregnancy and are related to negative outcomes for child development and health. Modifications in child DNA methylation are discussed as an underlying mechanism for the association between prenatal depressive symptoms and alterations in child outcomes. However, formerly reported genome-wide associations have yet to be replicated.</p><p><strong>Methods: </strong>In an epigenome-wide association study (EWAS), alterations of DNA methylation related to maternal prenatal depressive symptoms were investigated in buccal cell samples from 174 children (n = 52 exposed to prenatal depressive symptoms; 6-9 years old) of the German longitudinal study FRAMES-FRANCES. Whole blood samples from the independent, age-comparable ARIES subsample of the ARIES/ALSPAC study (n = 641; n = 159 exposed to prenatal depressive symptoms; 7-8 years old) were examined as a confirmation sample. Depressive symptoms were assessed with the Edinburgh Postnatal Depression Scale. DNA methylation was analyzed with the Infinium Human Methylation 450k BeadChip. Modifications in single CpGs, regions, and biological pathways were investigated. Results were adjusted for age and birth outcomes as well as postnatal and current maternal depressive symptoms. Analyses were performed for the whole sample as well as separated for sex.</p><p><strong>Results: </strong>The EWAS yielded no differentially methylated CpG or region as well as no accordance between samples withstanding correction for multiple testing. In pathway analyses, no overlapping functional domain was found to be enriched for either sample. A comparison of current and former findings suggests some overlapping methylation modifications from infancy to childhood. Results suggest that there might be sex-specific differential methylation, which should be further investigated in additional studies.</p><p><strong>Conclusions: </strong>The current, mainly nonsignificant, results challenge the assumption of consistent modifications of DNA methylation in children exposed to prenatal depressive symptoms. Despite the relatively small sample size used in this study, this lack of significant results may reflect diverse issues of environmental epigenetic studies, which need to be addressed in future research.</p>\",\"PeriodicalId\":41996,\"journal\":{\"name\":\"Epigenetics Insights\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2020-06-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/e5/0c/10.1177_2516865720932146.PMC7298426.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Epigenetics Insights\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/2516865720932146\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2020/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Epigenetics Insights","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/2516865720932146","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
背景:孕妇抑郁症状是孕期的常见现象,与儿童发育和健康的负面结果有关。儿童 DNA 甲基化的改变被认为是产前抑郁症状与儿童结局改变之间关联的潜在机制。然而,以前报道的全基因组关联尚未得到证实:在一项全表观基因组关联研究(EWAS)中,研究人员对德国纵向研究 FRAMES-FRANCES 中 174 名儿童(n = 52 名暴露于产前抑郁症状;6-9 岁)的口腔细胞样本中与母亲产前抑郁症状相关的 DNA 甲基化改变进行了调查。ARIES/ALSPAC研究中独立的、具有年龄可比性的ARIES子样本(n = 641;n = 159暴露于产前抑郁症状;7-8岁)的全血样本作为确认样本进行了检测。抑郁症状采用爱丁堡产后抑郁量表进行评估。DNA 甲基化用 Infinium Human Methylation 450k BeadChip 进行分析。调查了单个 CpGs、区域和生物通路的甲基化情况。研究结果根据年龄、出生结果以及产后和当前产妇抑郁症状进行了调整。对整个样本以及按性别分列的样本进行了分析:结果:EWAS 没有发现差异甲基化的 CpG 或区域,样本间也没有经得起多重检验校正的差异。在通路分析中,两个样本都没有发现重叠功能域的富集。将目前的研究结果与以前的研究结果进行比较后发现,从婴儿期到儿童期,甲基化修饰存在一些重叠。结果表明,可能存在性别特异性甲基化差异,这需要在其他研究中进一步调查:结论:目前的研究结果主要是非显著性的,这些结果对产前有抑郁症状的儿童 DNA 甲基化发生一致改变的假设提出了质疑。尽管本研究使用的样本量相对较小,但缺乏显著结果可能反映了环境表观遗传研究中的各种问题,需要在今后的研究中加以解决。
Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts.
Background: Maternal depressive symptoms are a common phenomenon during pregnancy and are related to negative outcomes for child development and health. Modifications in child DNA methylation are discussed as an underlying mechanism for the association between prenatal depressive symptoms and alterations in child outcomes. However, formerly reported genome-wide associations have yet to be replicated.
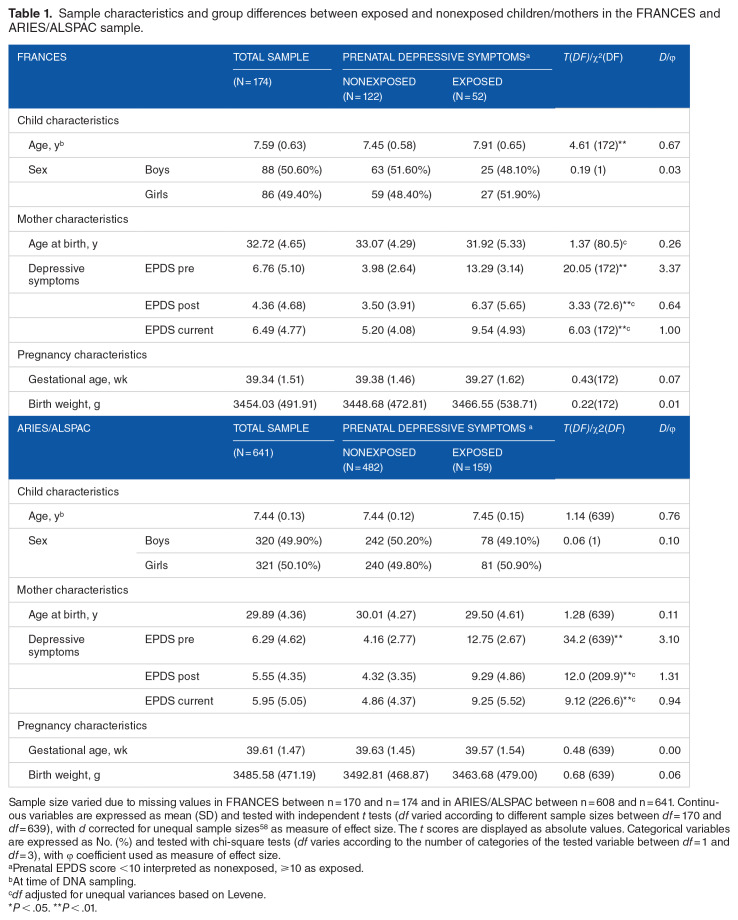
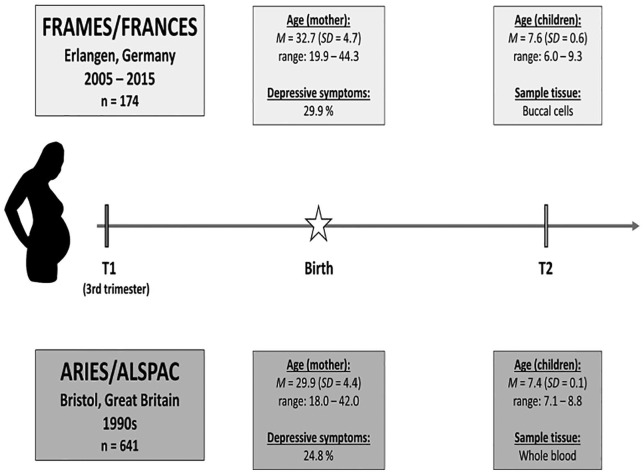
Methods: In an epigenome-wide association study (EWAS), alterations of DNA methylation related to maternal prenatal depressive symptoms were investigated in buccal cell samples from 174 children (n = 52 exposed to prenatal depressive symptoms; 6-9 years old) of the German longitudinal study FRAMES-FRANCES. Whole blood samples from the independent, age-comparable ARIES subsample of the ARIES/ALSPAC study (n = 641; n = 159 exposed to prenatal depressive symptoms; 7-8 years old) were examined as a confirmation sample. Depressive symptoms were assessed with the Edinburgh Postnatal Depression Scale. DNA methylation was analyzed with the Infinium Human Methylation 450k BeadChip. Modifications in single CpGs, regions, and biological pathways were investigated. Results were adjusted for age and birth outcomes as well as postnatal and current maternal depressive symptoms. Analyses were performed for the whole sample as well as separated for sex.
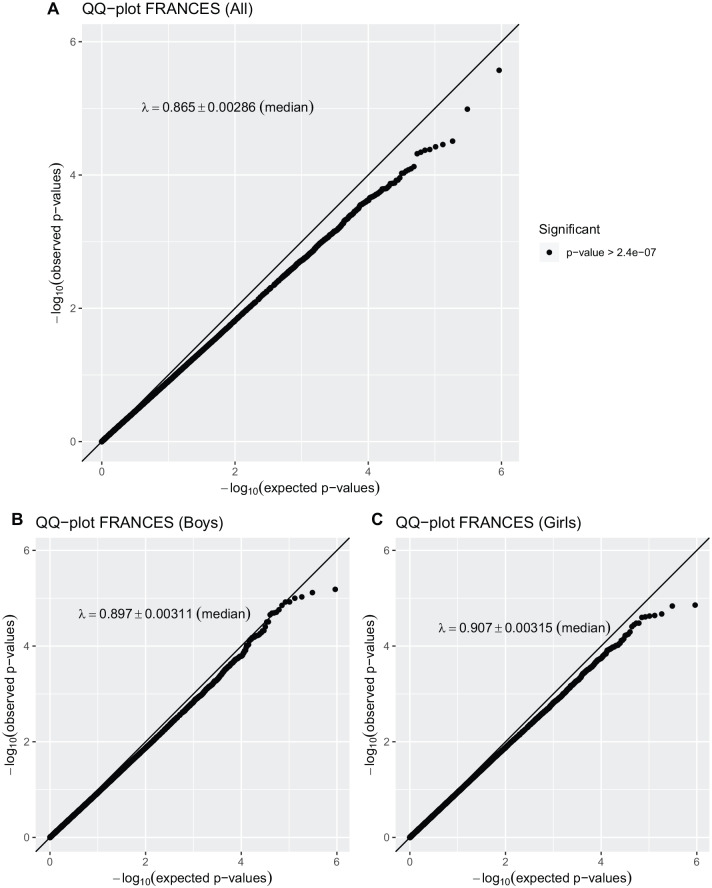
Results: The EWAS yielded no differentially methylated CpG or region as well as no accordance between samples withstanding correction for multiple testing. In pathway analyses, no overlapping functional domain was found to be enriched for either sample. A comparison of current and former findings suggests some overlapping methylation modifications from infancy to childhood. Results suggest that there might be sex-specific differential methylation, which should be further investigated in additional studies.
Conclusions: The current, mainly nonsignificant, results challenge the assumption of consistent modifications of DNA methylation in children exposed to prenatal depressive symptoms. Despite the relatively small sample size used in this study, this lack of significant results may reflect diverse issues of environmental epigenetic studies, which need to be addressed in future research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


