{"title":"脊髓小脑性共济失调23型的家族内表型变异。","authors":"Shunichi Satoh, Yasufumi Kondo, Shinji Ohara, Tomomi Yamaguchi, Katsuya Nakamura, Kunihiro Yoshida","doi":"10.1186/s40673-020-00117-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Spinocerebellar ataxia type 23 (SCA23) is an autosomal dominant cerebellar ataxia caused by pathogenic variants in the prodynorphin gene (<i>PDYN</i>). The frequency of <i>PDYN</i> variants is reportedly very low (~ 0.1%) in several ataxia cohorts screened to date.</p><p><strong>Case presentations: </strong>We found five cases of SCA23 in two families (mean age at onset: 37.8 ± 5.5 years; mean age at examination: 64.2 ± 12.3 years) with a novel <i>PDYN</i> variant (c.644G > A:p.R215H). We identified marked heterogeneity in the clinical features in Family 1: the proband showed clinical and neuroimaging features suggestive of multiple system atrophy with predominant parkinsonism (MSA-P). Conversely, the proband's mother with the <i>PDYN</i> p.R215H variant had no subjective symptoms; she had not come to medical attention before our survey, although she showed apparent cerebellar atrophy on brain magnetic resonance imaging (MRI). The other two patients in Family 1 and a patient in Family 2 showed slowly progressive cerebellar ataxia.</p><p><strong>Conclusions: </strong>We here report two Japanese families with SCA23, one of which showed considerable phenotypic variation in affected members. Our findings support that SCA23 can phenotypically overlap with MSA.</p>","PeriodicalId":36752,"journal":{"name":"Cerebellum and Ataxias","volume":"7 ","pages":"7"},"PeriodicalIF":0.0000,"publicationDate":"2020-06-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s40673-020-00117-x","citationCount":"2","resultStr":"{\"title\":\"Intrafamilial phenotypic variation in spinocerebellar ataxia type 23.\",\"authors\":\"Shunichi Satoh, Yasufumi Kondo, Shinji Ohara, Tomomi Yamaguchi, Katsuya Nakamura, Kunihiro Yoshida\",\"doi\":\"10.1186/s40673-020-00117-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Spinocerebellar ataxia type 23 (SCA23) is an autosomal dominant cerebellar ataxia caused by pathogenic variants in the prodynorphin gene (<i>PDYN</i>). The frequency of <i>PDYN</i> variants is reportedly very low (~ 0.1%) in several ataxia cohorts screened to date.</p><p><strong>Case presentations: </strong>We found five cases of SCA23 in two families (mean age at onset: 37.8 ± 5.5 years; mean age at examination: 64.2 ± 12.3 years) with a novel <i>PDYN</i> variant (c.644G > A:p.R215H). We identified marked heterogeneity in the clinical features in Family 1: the proband showed clinical and neuroimaging features suggestive of multiple system atrophy with predominant parkinsonism (MSA-P). Conversely, the proband's mother with the <i>PDYN</i> p.R215H variant had no subjective symptoms; she had not come to medical attention before our survey, although she showed apparent cerebellar atrophy on brain magnetic resonance imaging (MRI). The other two patients in Family 1 and a patient in Family 2 showed slowly progressive cerebellar ataxia.</p><p><strong>Conclusions: </strong>We here report two Japanese families with SCA23, one of which showed considerable phenotypic variation in affected members. Our findings support that SCA23 can phenotypically overlap with MSA.</p>\",\"PeriodicalId\":36752,\"journal\":{\"name\":\"Cerebellum and Ataxias\",\"volume\":\"7 \",\"pages\":\"7\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-06-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s40673-020-00117-x\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cerebellum and Ataxias\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s40673-020-00117-x\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2020/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cerebellum and Ataxias","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40673-020-00117-x","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
Intrafamilial phenotypic variation in spinocerebellar ataxia type 23.
Background: Spinocerebellar ataxia type 23 (SCA23) is an autosomal dominant cerebellar ataxia caused by pathogenic variants in the prodynorphin gene (PDYN). The frequency of PDYN variants is reportedly very low (~ 0.1%) in several ataxia cohorts screened to date.
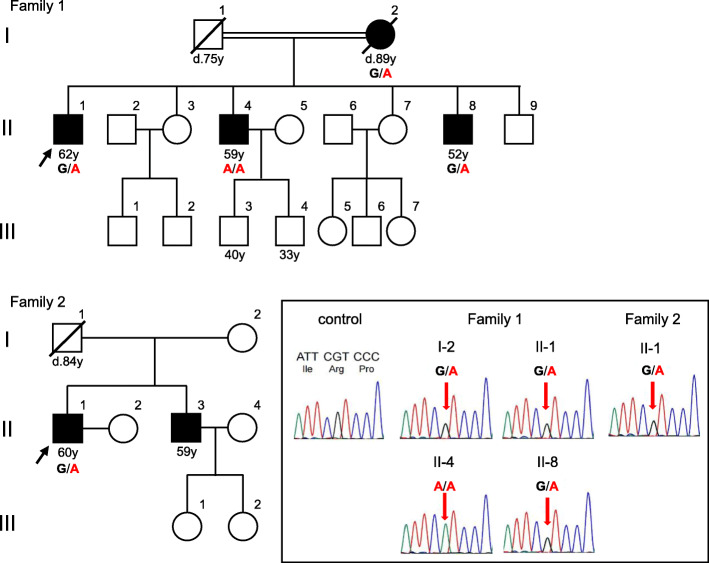
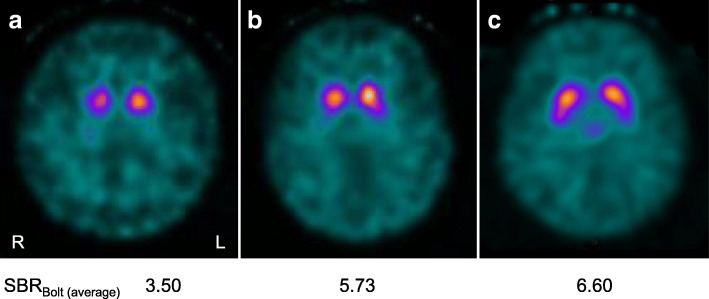
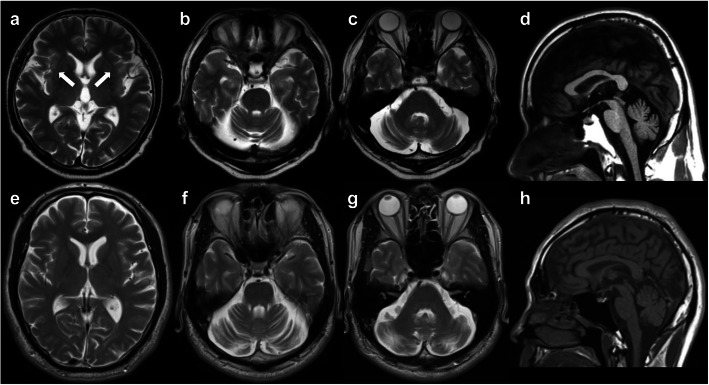
Case presentations: We found five cases of SCA23 in two families (mean age at onset: 37.8 ± 5.5 years; mean age at examination: 64.2 ± 12.3 years) with a novel PDYN variant (c.644G > A:p.R215H). We identified marked heterogeneity in the clinical features in Family 1: the proband showed clinical and neuroimaging features suggestive of multiple system atrophy with predominant parkinsonism (MSA-P). Conversely, the proband's mother with the PDYN p.R215H variant had no subjective symptoms; she had not come to medical attention before our survey, although she showed apparent cerebellar atrophy on brain magnetic resonance imaging (MRI). The other two patients in Family 1 and a patient in Family 2 showed slowly progressive cerebellar ataxia.
Conclusions: We here report two Japanese families with SCA23, one of which showed considerable phenotypic variation in affected members. Our findings support that SCA23 can phenotypically overlap with MSA.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


