Amirmasoud Lanjan, Zahra Moradi and Seshasai Srinivasan
{"title":"应用量子力学评价分子动力学势参数的计算框架[j]","authors":"Amirmasoud Lanjan, Zahra Moradi and Seshasai Srinivasan","doi":"10.1039/D3ME00007A","DOIUrl":null,"url":null,"abstract":"<p >Molecular dynamics (MD) and quantum mechanics (QM) calculations can be used to characterize novel materials and phenomena that experimental methods cannot capture. While QM provides accurate results, it has high computational costs and is applicable only to small system sizes. On the other hand, MD can work with larger systems and has better computational efficiency but is incapable of studying novel materials/phenomena due to its dependency on experimental data in the literature. Therefore, complex systems such as solid–electrolyte interface (SEI) layer formation cannot be comprehensively investigated by (I) experimental methods due to small time scales, (II) MD simulations because of the absence of experimental data, and (III) QM calculations due to the relatively large system. Herein, we report a suite of new nano-scale algorithms to facilitate studying complex material interphases and molecular systems with the accuracy and precision of QM calculations and at a speed and system size permissible using MD simulations. Our formulation addresses the most challenging aspect of performing an MD simulation, <em>i.e.</em>, finding accurate potential (force field) parameters that are often derived from experimental methods. The computational framework presented in this work consists of seven main functions/algorithms that collectively help us account for the effects of nonbonded, bonded, angle, dihedral, and improper interactions in a system/molecule. It is now possible to use these simulations to design and study wholly new and novel materials and investigate phenomena at an atomic/molecular scale under different conditions without the need for prior experimental investigations. We have successfully validated our algorithms with respect to the experimental data of established materials such as H<small><sub>2</sub></small>O (a polar molecule), LiPF<small><sub>6</sub></small> (an ionic compound), C<small><sub>2</sub></small>H<small><sub>5</sub></small>OH (ethanol), C<small><sub>8</sub></small>H<small><sub>18</sub></small> (a long chain molecule), and ethylene carbonate (EC) (a complex molecular system). The obtained results have an accuracy of over 90%.</p>","PeriodicalId":91,"journal":{"name":"Molecular Systems Design & Engineering","volume":" 5","pages":" 632-646"},"PeriodicalIF":3.2000,"publicationDate":"2023-02-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"2","resultStr":"{\"title\":\"A computational framework for evaluating molecular dynamics potential parameters employing quantum mechanics†\",\"authors\":\"Amirmasoud Lanjan, Zahra Moradi and Seshasai Srinivasan\",\"doi\":\"10.1039/D3ME00007A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Molecular dynamics (MD) and quantum mechanics (QM) calculations can be used to characterize novel materials and phenomena that experimental methods cannot capture. While QM provides accurate results, it has high computational costs and is applicable only to small system sizes. On the other hand, MD can work with larger systems and has better computational efficiency but is incapable of studying novel materials/phenomena due to its dependency on experimental data in the literature. Therefore, complex systems such as solid–electrolyte interface (SEI) layer formation cannot be comprehensively investigated by (I) experimental methods due to small time scales, (II) MD simulations because of the absence of experimental data, and (III) QM calculations due to the relatively large system. Herein, we report a suite of new nano-scale algorithms to facilitate studying complex material interphases and molecular systems with the accuracy and precision of QM calculations and at a speed and system size permissible using MD simulations. Our formulation addresses the most challenging aspect of performing an MD simulation, <em>i.e.</em>, finding accurate potential (force field) parameters that are often derived from experimental methods. The computational framework presented in this work consists of seven main functions/algorithms that collectively help us account for the effects of nonbonded, bonded, angle, dihedral, and improper interactions in a system/molecule. It is now possible to use these simulations to design and study wholly new and novel materials and investigate phenomena at an atomic/molecular scale under different conditions without the need for prior experimental investigations. We have successfully validated our algorithms with respect to the experimental data of established materials such as H<small><sub>2</sub></small>O (a polar molecule), LiPF<small><sub>6</sub></small> (an ionic compound), C<small><sub>2</sub></small>H<small><sub>5</sub></small>OH (ethanol), C<small><sub>8</sub></small>H<small><sub>18</sub></small> (a long chain molecule), and ethylene carbonate (EC) (a complex molecular system). The obtained results have an accuracy of over 90%.</p>\",\"PeriodicalId\":91,\"journal\":{\"name\":\"Molecular Systems Design & Engineering\",\"volume\":\" 5\",\"pages\":\" 632-646\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2023-02-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Systems Design & Engineering\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2023/me/d3me00007a\",\"RegionNum\":3,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Design & Engineering","FirstCategoryId":"5","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2023/me/d3me00007a","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
A computational framework for evaluating molecular dynamics potential parameters employing quantum mechanics†
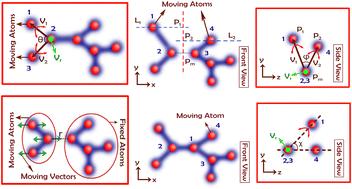
Molecular dynamics (MD) and quantum mechanics (QM) calculations can be used to characterize novel materials and phenomena that experimental methods cannot capture. While QM provides accurate results, it has high computational costs and is applicable only to small system sizes. On the other hand, MD can work with larger systems and has better computational efficiency but is incapable of studying novel materials/phenomena due to its dependency on experimental data in the literature. Therefore, complex systems such as solid–electrolyte interface (SEI) layer formation cannot be comprehensively investigated by (I) experimental methods due to small time scales, (II) MD simulations because of the absence of experimental data, and (III) QM calculations due to the relatively large system. Herein, we report a suite of new nano-scale algorithms to facilitate studying complex material interphases and molecular systems with the accuracy and precision of QM calculations and at a speed and system size permissible using MD simulations. Our formulation addresses the most challenging aspect of performing an MD simulation, i.e., finding accurate potential (force field) parameters that are often derived from experimental methods. The computational framework presented in this work consists of seven main functions/algorithms that collectively help us account for the effects of nonbonded, bonded, angle, dihedral, and improper interactions in a system/molecule. It is now possible to use these simulations to design and study wholly new and novel materials and investigate phenomena at an atomic/molecular scale under different conditions without the need for prior experimental investigations. We have successfully validated our algorithms with respect to the experimental data of established materials such as H2O (a polar molecule), LiPF6 (an ionic compound), C2H5OH (ethanol), C8H18 (a long chain molecule), and ethylene carbonate (EC) (a complex molecular system). The obtained results have an accuracy of over 90%.
期刊介绍:
Molecular Systems Design & Engineering provides a hub for cutting-edge research into how understanding of molecular properties, behaviour and interactions can be used to design and assemble better materials, systems, and processes to achieve specific functions. These may have applications of technological significance and help address global challenges.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


