Tzu-Hsuan Chao, Shiv Rekhi, Jeetain Mittal and Daniel P. Tabor
{"title":"数据驱动模型预测本质上无序的蛋白质聚合物物理直接从组成或序列†","authors":"Tzu-Hsuan Chao, Shiv Rekhi, Jeetain Mittal and Daniel P. Tabor","doi":"10.1039/D3ME00053B","DOIUrl":null,"url":null,"abstract":"<p >The molecular-level understanding of intrinsically disordered proteins is challenging due to experimental characterization difficulties. Computational understanding of IDPs also requires fundamental advances, as the leading tools for predicting protein folding (<em>e.g.</em>, AlphaFold), typically fail to describe the structural ensembles of IDPs. The focus of this paper is to 1) develop new representations for intrinsically disordered proteins and 2) pair these representations with classical machine learning and deep learning models to predict the radius of gyration and derived scaling exponent of IDPs. Here, we build a new physically-motivated feature called the bag of amino acid interactions representation, which encodes pairwise interactions explicitly into the representation. This feature essentially counts and weights all possible non-bonded interactions in a sequence and thus is, in principle, compatible with arbitrary sequence lengths. To see how well this new feature performs, both categorical and physically-motivated featurization techniques are tested on a computational dataset containing 10 000 sequences simulated at the coarse-grained level. The results indicate that this new feature outperforms the other purely categorical and physically-motivated features and possesses solid extrapolation capabilities. For future use, this feature can potentially provide physical insights into amino acid interactions, including their temperature dependence, and be applied to other protein spaces.</p>","PeriodicalId":91,"journal":{"name":"Molecular Systems Design & Engineering","volume":" 9","pages":" 1146-1155"},"PeriodicalIF":3.2000,"publicationDate":"2023-06-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"3","resultStr":"{\"title\":\"Data-driven models for predicting intrinsically disordered protein polymer physics directly from composition or sequence†\",\"authors\":\"Tzu-Hsuan Chao, Shiv Rekhi, Jeetain Mittal and Daniel P. Tabor\",\"doi\":\"10.1039/D3ME00053B\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The molecular-level understanding of intrinsically disordered proteins is challenging due to experimental characterization difficulties. Computational understanding of IDPs also requires fundamental advances, as the leading tools for predicting protein folding (<em>e.g.</em>, AlphaFold), typically fail to describe the structural ensembles of IDPs. The focus of this paper is to 1) develop new representations for intrinsically disordered proteins and 2) pair these representations with classical machine learning and deep learning models to predict the radius of gyration and derived scaling exponent of IDPs. Here, we build a new physically-motivated feature called the bag of amino acid interactions representation, which encodes pairwise interactions explicitly into the representation. This feature essentially counts and weights all possible non-bonded interactions in a sequence and thus is, in principle, compatible with arbitrary sequence lengths. To see how well this new feature performs, both categorical and physically-motivated featurization techniques are tested on a computational dataset containing 10 000 sequences simulated at the coarse-grained level. The results indicate that this new feature outperforms the other purely categorical and physically-motivated features and possesses solid extrapolation capabilities. For future use, this feature can potentially provide physical insights into amino acid interactions, including their temperature dependence, and be applied to other protein spaces.</p>\",\"PeriodicalId\":91,\"journal\":{\"name\":\"Molecular Systems Design & Engineering\",\"volume\":\" 9\",\"pages\":\" 1146-1155\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2023-06-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Systems Design & Engineering\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2023/me/d3me00053b\",\"RegionNum\":3,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Design & Engineering","FirstCategoryId":"5","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2023/me/d3me00053b","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Data-driven models for predicting intrinsically disordered protein polymer physics directly from composition or sequence†
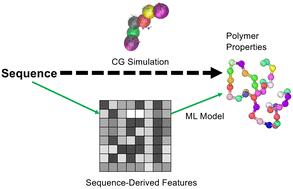
The molecular-level understanding of intrinsically disordered proteins is challenging due to experimental characterization difficulties. Computational understanding of IDPs also requires fundamental advances, as the leading tools for predicting protein folding (e.g., AlphaFold), typically fail to describe the structural ensembles of IDPs. The focus of this paper is to 1) develop new representations for intrinsically disordered proteins and 2) pair these representations with classical machine learning and deep learning models to predict the radius of gyration and derived scaling exponent of IDPs. Here, we build a new physically-motivated feature called the bag of amino acid interactions representation, which encodes pairwise interactions explicitly into the representation. This feature essentially counts and weights all possible non-bonded interactions in a sequence and thus is, in principle, compatible with arbitrary sequence lengths. To see how well this new feature performs, both categorical and physically-motivated featurization techniques are tested on a computational dataset containing 10 000 sequences simulated at the coarse-grained level. The results indicate that this new feature outperforms the other purely categorical and physically-motivated features and possesses solid extrapolation capabilities. For future use, this feature can potentially provide physical insights into amino acid interactions, including their temperature dependence, and be applied to other protein spaces.
期刊介绍:
Molecular Systems Design & Engineering provides a hub for cutting-edge research into how understanding of molecular properties, behaviour and interactions can be used to design and assemble better materials, systems, and processes to achieve specific functions. These may have applications of technological significance and help address global challenges.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


