{"title":"利用Huber罚凸优化函数†从基因表达数据预测代谢通量","authors":"Shao-Wu Zhang, Wang-Long Gou and Yan Li","doi":"10.1039/C6MB00811A","DOIUrl":null,"url":null,"abstract":"<p >As one of the critical parameters of a metabolic pathway, the metabolic flux in a metabolic network serves as an essential role in physiology and pathology. Constraint-based metabolic models are the widely used frameworks for predicting metabolic fluxes in genome-scale metabolic networks. Integrating the transcriptomic data into the constraint-based metabolic models can effectively predict context-specific fluxes across different conditions. However, these methods always need user-defined thresholds to identify the expression levels of metabolic genes or restrain the rate of biomass production, and the predictive results are sensitive to the thresholds. In this work, we present the Huber penalty convex optimization function (HPCOF) combined with the flux minimization principle to predict metabolic fluxes. Our HPCOF method integrates gene expression profiles into the genome-scale metabolic models (GEMs) to reduce the sensitivity to outliers, and uses continuous expression data to avoid selection of arbitrary threshold parameters. In the case studies of <em>Saccharomyces cerevisiae</em> (<em>S. cerevisiae</em>) and <em>Escherichia coli</em> (<em>E. coli</em>) strains under different conditions, the results show that our HPCOF method has a better fit to the experimentally measured values, and has a higher Pearson correlation coefficient, a smaller <em>P</em>-value and a lower sum of squared error than other methods. The HPCOF code can be freely downloaded from https://github.com/nwpu903/HPCOF for academic users.</p>","PeriodicalId":90,"journal":{"name":"Molecular BioSystems","volume":" 5","pages":" 901-909"},"PeriodicalIF":3.7430,"publicationDate":"2017-03-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1039/C6MB00811A","citationCount":"11","resultStr":"{\"title\":\"Prediction of metabolic fluxes from gene expression data with Huber penalty convex optimization function†\",\"authors\":\"Shao-Wu Zhang, Wang-Long Gou and Yan Li\",\"doi\":\"10.1039/C6MB00811A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >As one of the critical parameters of a metabolic pathway, the metabolic flux in a metabolic network serves as an essential role in physiology and pathology. Constraint-based metabolic models are the widely used frameworks for predicting metabolic fluxes in genome-scale metabolic networks. Integrating the transcriptomic data into the constraint-based metabolic models can effectively predict context-specific fluxes across different conditions. However, these methods always need user-defined thresholds to identify the expression levels of metabolic genes or restrain the rate of biomass production, and the predictive results are sensitive to the thresholds. In this work, we present the Huber penalty convex optimization function (HPCOF) combined with the flux minimization principle to predict metabolic fluxes. Our HPCOF method integrates gene expression profiles into the genome-scale metabolic models (GEMs) to reduce the sensitivity to outliers, and uses continuous expression data to avoid selection of arbitrary threshold parameters. In the case studies of <em>Saccharomyces cerevisiae</em> (<em>S. cerevisiae</em>) and <em>Escherichia coli</em> (<em>E. coli</em>) strains under different conditions, the results show that our HPCOF method has a better fit to the experimentally measured values, and has a higher Pearson correlation coefficient, a smaller <em>P</em>-value and a lower sum of squared error than other methods. The HPCOF code can be freely downloaded from https://github.com/nwpu903/HPCOF for academic users.</p>\",\"PeriodicalId\":90,\"journal\":{\"name\":\"Molecular BioSystems\",\"volume\":\" 5\",\"pages\":\" 901-909\"},\"PeriodicalIF\":3.7430,\"publicationDate\":\"2017-03-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1039/C6MB00811A\",\"citationCount\":\"11\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular BioSystems\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2017/mb/c6mb00811a\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular BioSystems","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2017/mb/c6mb00811a","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Prediction of metabolic fluxes from gene expression data with Huber penalty convex optimization function†
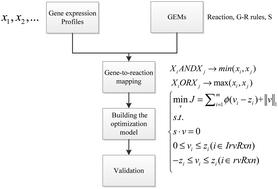
As one of the critical parameters of a metabolic pathway, the metabolic flux in a metabolic network serves as an essential role in physiology and pathology. Constraint-based metabolic models are the widely used frameworks for predicting metabolic fluxes in genome-scale metabolic networks. Integrating the transcriptomic data into the constraint-based metabolic models can effectively predict context-specific fluxes across different conditions. However, these methods always need user-defined thresholds to identify the expression levels of metabolic genes or restrain the rate of biomass production, and the predictive results are sensitive to the thresholds. In this work, we present the Huber penalty convex optimization function (HPCOF) combined with the flux minimization principle to predict metabolic fluxes. Our HPCOF method integrates gene expression profiles into the genome-scale metabolic models (GEMs) to reduce the sensitivity to outliers, and uses continuous expression data to avoid selection of arbitrary threshold parameters. In the case studies of Saccharomyces cerevisiae (S. cerevisiae) and Escherichia coli (E. coli) strains under different conditions, the results show that our HPCOF method has a better fit to the experimentally measured values, and has a higher Pearson correlation coefficient, a smaller P-value and a lower sum of squared error than other methods. The HPCOF code can be freely downloaded from https://github.com/nwpu903/HPCOF for academic users.
期刊介绍:
Molecular Omics publishes molecular level experimental and bioinformatics research in the -omics sciences, including genomics, proteomics, transcriptomics and metabolomics. We will also welcome multidisciplinary papers presenting studies combining different types of omics, or the interface of omics and other fields such as systems biology or chemical biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


