Timothy R. Rudd, Mark D. Preston and Edwin A. Yates
{"title":"通过网络分析确定了437种肝素结合蛋白中保守碱性氨基酸序列的性质","authors":"Timothy R. Rudd, Mark D. Preston and Edwin A. Yates","doi":"10.1039/C6MB00857G","DOIUrl":null,"url":null,"abstract":"<p >In multicellular organisms, a large number of proteins interact with the polyanionic polysaccharides heparan sulphate (HS) and heparin. These interactions are usually assumed to be dominated by charge–charge interactions between the anionic carboxylate and/or sulfate groups of the polysaccharide and cationic amino acids of the protein. A major question is whether there exist conserved amino acid sequences for HS/heparin binding among these diverse proteins. Potentially conserved HS/heparin binding sequences were sought amongst 437 HS/heparin binding proteins. Amino acid sequences were extracted and compared using a <em>Levenshtein</em> distance metric. The resultant similarity matrices were visualised as graphs, enabling extraction of strongly conserved sequences from highly variable primary sequences while excluding short, core regions. This approach did not reveal extensive, conserved HS/heparin binding sequences, rather a number of shorter, more widely spaced sequences that may work in unison to form heparin-binding sites on protein surfaces, arguing for convergent evolution. Thus, it is the three-dimensional arrangement of these conserved motifs on the protein surface, rather than the primary sequence <em>per se</em>, which are the evolutionary elements.</p>","PeriodicalId":90,"journal":{"name":"Molecular BioSystems","volume":" 5","pages":" 852-865"},"PeriodicalIF":3.7430,"publicationDate":"2017-03-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1039/C6MB00857G","citationCount":"31","resultStr":"{\"title\":\"The nature of the conserved basic amino acid sequences found among 437 heparin binding proteins determined by network analysis†\",\"authors\":\"Timothy R. Rudd, Mark D. Preston and Edwin A. Yates\",\"doi\":\"10.1039/C6MB00857G\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In multicellular organisms, a large number of proteins interact with the polyanionic polysaccharides heparan sulphate (HS) and heparin. These interactions are usually assumed to be dominated by charge–charge interactions between the anionic carboxylate and/or sulfate groups of the polysaccharide and cationic amino acids of the protein. A major question is whether there exist conserved amino acid sequences for HS/heparin binding among these diverse proteins. Potentially conserved HS/heparin binding sequences were sought amongst 437 HS/heparin binding proteins. Amino acid sequences were extracted and compared using a <em>Levenshtein</em> distance metric. The resultant similarity matrices were visualised as graphs, enabling extraction of strongly conserved sequences from highly variable primary sequences while excluding short, core regions. This approach did not reveal extensive, conserved HS/heparin binding sequences, rather a number of shorter, more widely spaced sequences that may work in unison to form heparin-binding sites on protein surfaces, arguing for convergent evolution. Thus, it is the three-dimensional arrangement of these conserved motifs on the protein surface, rather than the primary sequence <em>per se</em>, which are the evolutionary elements.</p>\",\"PeriodicalId\":90,\"journal\":{\"name\":\"Molecular BioSystems\",\"volume\":\" 5\",\"pages\":\" 852-865\"},\"PeriodicalIF\":3.7430,\"publicationDate\":\"2017-03-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1039/C6MB00857G\",\"citationCount\":\"31\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular BioSystems\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2017/mb/c6mb00857g\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular BioSystems","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2017/mb/c6mb00857g","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
The nature of the conserved basic amino acid sequences found among 437 heparin binding proteins determined by network analysis†
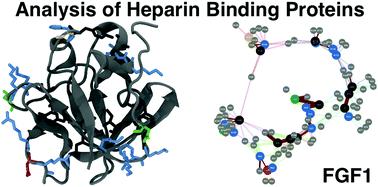
In multicellular organisms, a large number of proteins interact with the polyanionic polysaccharides heparan sulphate (HS) and heparin. These interactions are usually assumed to be dominated by charge–charge interactions between the anionic carboxylate and/or sulfate groups of the polysaccharide and cationic amino acids of the protein. A major question is whether there exist conserved amino acid sequences for HS/heparin binding among these diverse proteins. Potentially conserved HS/heparin binding sequences were sought amongst 437 HS/heparin binding proteins. Amino acid sequences were extracted and compared using a Levenshtein distance metric. The resultant similarity matrices were visualised as graphs, enabling extraction of strongly conserved sequences from highly variable primary sequences while excluding short, core regions. This approach did not reveal extensive, conserved HS/heparin binding sequences, rather a number of shorter, more widely spaced sequences that may work in unison to form heparin-binding sites on protein surfaces, arguing for convergent evolution. Thus, it is the three-dimensional arrangement of these conserved motifs on the protein surface, rather than the primary sequence per se, which are the evolutionary elements.
期刊介绍:
Molecular Omics publishes molecular level experimental and bioinformatics research in the -omics sciences, including genomics, proteomics, transcriptomics and metabolomics. We will also welcome multidisciplinary papers presenting studies combining different types of omics, or the interface of omics and other fields such as systems biology or chemical biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


