Tina Jenewein, Scott A Kanner, Daniel Bauer, Brigitte Hertel, Henry M Colecraft, Anna Moroni, Gerhard Thiel, Silke Kauferstein
{"title":"hERG钾通道PAS结构域L69P突变导致转运不足导致LQTS。","authors":"Tina Jenewein, Scott A Kanner, Daniel Bauer, Brigitte Hertel, Henry M Colecraft, Anna Moroni, Gerhard Thiel, Silke Kauferstein","doi":"10.1080/19336950.2020.1751522","DOIUrl":null,"url":null,"abstract":"<p><p>The congenital long QT syndrome (LQTS) is a cardiac disorder characterized by a prolonged QT interval on the electrocardiogram and an increased susceptibility to ventricular arrhythmias and sudden cardiac death. A frequent cause for LQTS is mutations in the <i>KCNH2</i> gene (also known as the <i>human ether-a-go-go-related gene</i> or <i>hERG</i>), which reduce or modulate the potassium current I<sub>Kr</sub> and hence alter cardiac repolarization. In a patient with a clinically diagnosed LQTS, we identified the mutation L69P in the N-terminal PAS (Per-Arnt-Sim) domain of hERG. Functional expression in HEK293 cells shows that a homotetrameric hERG channel reconstituted with only mutant subunits exhibits a drastically reduced surface expression of the channel protein thus leading to a diminished hERG current. Unlike many other mutations in the hERG-PAS domain the negative impact of the L69P substitution cannot be rescued by facilitated protein folding at a lower incubation temperature. Further, co-expression of wt and mutant monomers does not restore either wt like surface expression or the full hERG current. These results indicate L69P is a dominant negative mutation, with deficits which most likely occurs at the level of protein folding and subsequently inhibits trafficking to the plasma membrane. The functional deficits of the mutant channel support the clinical diagnosis of a LQTS.</p>","PeriodicalId":72555,"journal":{"name":"Channels (Austin, Tex.)","volume":null,"pages":null},"PeriodicalIF":0.0000,"publicationDate":"2020-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1080/19336950.2020.1751522","citationCount":"1","resultStr":"{\"title\":\"The mutation L69P in the PAS domain of the hERG potassium channel results in LQTS by trafficking deficiency.\",\"authors\":\"Tina Jenewein, Scott A Kanner, Daniel Bauer, Brigitte Hertel, Henry M Colecraft, Anna Moroni, Gerhard Thiel, Silke Kauferstein\",\"doi\":\"10.1080/19336950.2020.1751522\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The congenital long QT syndrome (LQTS) is a cardiac disorder characterized by a prolonged QT interval on the electrocardiogram and an increased susceptibility to ventricular arrhythmias and sudden cardiac death. A frequent cause for LQTS is mutations in the <i>KCNH2</i> gene (also known as the <i>human ether-a-go-go-related gene</i> or <i>hERG</i>), which reduce or modulate the potassium current I<sub>Kr</sub> and hence alter cardiac repolarization. In a patient with a clinically diagnosed LQTS, we identified the mutation L69P in the N-terminal PAS (Per-Arnt-Sim) domain of hERG. Functional expression in HEK293 cells shows that a homotetrameric hERG channel reconstituted with only mutant subunits exhibits a drastically reduced surface expression of the channel protein thus leading to a diminished hERG current. Unlike many other mutations in the hERG-PAS domain the negative impact of the L69P substitution cannot be rescued by facilitated protein folding at a lower incubation temperature. Further, co-expression of wt and mutant monomers does not restore either wt like surface expression or the full hERG current. These results indicate L69P is a dominant negative mutation, with deficits which most likely occurs at the level of protein folding and subsequently inhibits trafficking to the plasma membrane. The functional deficits of the mutant channel support the clinical diagnosis of a LQTS.</p>\",\"PeriodicalId\":72555,\"journal\":{\"name\":\"Channels (Austin, Tex.)\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1080/19336950.2020.1751522\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Channels (Austin, Tex.)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1080/19336950.2020.1751522\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Channels (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/19336950.2020.1751522","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
The mutation L69P in the PAS domain of the hERG potassium channel results in LQTS by trafficking deficiency.
The congenital long QT syndrome (LQTS) is a cardiac disorder characterized by a prolonged QT interval on the electrocardiogram and an increased susceptibility to ventricular arrhythmias and sudden cardiac death. A frequent cause for LQTS is mutations in the KCNH2 gene (also known as the human ether-a-go-go-related gene or hERG), which reduce or modulate the potassium current IKr and hence alter cardiac repolarization. In a patient with a clinically diagnosed LQTS, we identified the mutation L69P in the N-terminal PAS (Per-Arnt-Sim) domain of hERG. Functional expression in HEK293 cells shows that a homotetrameric hERG channel reconstituted with only mutant subunits exhibits a drastically reduced surface expression of the channel protein thus leading to a diminished hERG current. Unlike many other mutations in the hERG-PAS domain the negative impact of the L69P substitution cannot be rescued by facilitated protein folding at a lower incubation temperature. Further, co-expression of wt and mutant monomers does not restore either wt like surface expression or the full hERG current. These results indicate L69P is a dominant negative mutation, with deficits which most likely occurs at the level of protein folding and subsequently inhibits trafficking to the plasma membrane. The functional deficits of the mutant channel support the clinical diagnosis of a LQTS.
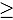

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


