Suttipong Suttapitugsakul, Haopeng Xiao, Johanna Smeekens and Ronghu Wu
{"title":"评价和优化还原和烷基化方法,以最大限度地利用MS-based蛋白质组学鉴定多肽","authors":"Suttipong Suttapitugsakul, Haopeng Xiao, Johanna Smeekens and Ronghu Wu","doi":"10.1039/C7MB00393E","DOIUrl":null,"url":null,"abstract":"<p >Mass spectrometry (MS) has become an increasingly important technique to analyze proteins. In popular bottom-up MS-based proteomics, reduction and alkylation are routine steps to facilitate peptide identification. However, incomplete reactions and side reactions may occur, which compromise the experimental results. In this work, we systematically evaluated the reduction step with commonly used reagents, <em>i.e.</em>, dithiothreitol, 2-mercaptoethanol, tris(2-carboxyethyl)phosphine, or tris(3-hydroxypropyl)phosphine, and alkylation with iodoacetamide, acrylamide, <em>N</em>-ethylmaleimide, or 4-vinylpyridine. By using digested peptides from a yeast whole-cell lysate, the number of proteins and peptides identified were very similar using four different reducing reagents. The results from four alkylating reagents, however, were dramatically different with iodoacetamide giving the highest number of peptides with alkylated cysteine and the lowest number of peptides with incomplete cysteine alkylation and side reactions. Alkylation conditions with iodoacetamide were further optimized. To identify more peptides with cysteine, thiopropyl-sepharose 6B resins were used to enrich them, and the optimal conditions were employed for the reduction and alkylation. The enrichment resulted in over three times more cysteine-containing peptides than without enrichment. Systematic evaluation of the reduction and alkylation with different reagents can aid in a better design of bottom-up proteomic experiments.</p>","PeriodicalId":90,"journal":{"name":"Molecular BioSystems","volume":" 12","pages":" 2574-2582"},"PeriodicalIF":3.7430,"publicationDate":"2017-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1039/C7MB00393E","citationCount":"47","resultStr":"{\"title\":\"Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics†‡\",\"authors\":\"Suttipong Suttapitugsakul, Haopeng Xiao, Johanna Smeekens and Ronghu Wu\",\"doi\":\"10.1039/C7MB00393E\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Mass spectrometry (MS) has become an increasingly important technique to analyze proteins. In popular bottom-up MS-based proteomics, reduction and alkylation are routine steps to facilitate peptide identification. However, incomplete reactions and side reactions may occur, which compromise the experimental results. In this work, we systematically evaluated the reduction step with commonly used reagents, <em>i.e.</em>, dithiothreitol, 2-mercaptoethanol, tris(2-carboxyethyl)phosphine, or tris(3-hydroxypropyl)phosphine, and alkylation with iodoacetamide, acrylamide, <em>N</em>-ethylmaleimide, or 4-vinylpyridine. By using digested peptides from a yeast whole-cell lysate, the number of proteins and peptides identified were very similar using four different reducing reagents. The results from four alkylating reagents, however, were dramatically different with iodoacetamide giving the highest number of peptides with alkylated cysteine and the lowest number of peptides with incomplete cysteine alkylation and side reactions. Alkylation conditions with iodoacetamide were further optimized. To identify more peptides with cysteine, thiopropyl-sepharose 6B resins were used to enrich them, and the optimal conditions were employed for the reduction and alkylation. The enrichment resulted in over three times more cysteine-containing peptides than without enrichment. Systematic evaluation of the reduction and alkylation with different reagents can aid in a better design of bottom-up proteomic experiments.</p>\",\"PeriodicalId\":90,\"journal\":{\"name\":\"Molecular BioSystems\",\"volume\":\" 12\",\"pages\":\" 2574-2582\"},\"PeriodicalIF\":3.7430,\"publicationDate\":\"2017-09-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1039/C7MB00393E\",\"citationCount\":\"47\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular BioSystems\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2017/mb/c7mb00393e\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular BioSystems","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2017/mb/c7mb00393e","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics†‡
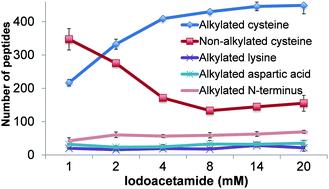
Mass spectrometry (MS) has become an increasingly important technique to analyze proteins. In popular bottom-up MS-based proteomics, reduction and alkylation are routine steps to facilitate peptide identification. However, incomplete reactions and side reactions may occur, which compromise the experimental results. In this work, we systematically evaluated the reduction step with commonly used reagents, i.e., dithiothreitol, 2-mercaptoethanol, tris(2-carboxyethyl)phosphine, or tris(3-hydroxypropyl)phosphine, and alkylation with iodoacetamide, acrylamide, N-ethylmaleimide, or 4-vinylpyridine. By using digested peptides from a yeast whole-cell lysate, the number of proteins and peptides identified were very similar using four different reducing reagents. The results from four alkylating reagents, however, were dramatically different with iodoacetamide giving the highest number of peptides with alkylated cysteine and the lowest number of peptides with incomplete cysteine alkylation and side reactions. Alkylation conditions with iodoacetamide were further optimized. To identify more peptides with cysteine, thiopropyl-sepharose 6B resins were used to enrich them, and the optimal conditions were employed for the reduction and alkylation. The enrichment resulted in over three times more cysteine-containing peptides than without enrichment. Systematic evaluation of the reduction and alkylation with different reagents can aid in a better design of bottom-up proteomic experiments.
期刊介绍:
Molecular Omics publishes molecular level experimental and bioinformatics research in the -omics sciences, including genomics, proteomics, transcriptomics and metabolomics. We will also welcome multidisciplinary papers presenting studies combining different types of omics, or the interface of omics and other fields such as systems biology or chemical biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


