Sudhir Srivastava, Michael Merchant, Anil Rai, Shesh N Rai
{"title":"标准化蛋白质组学工作流程的液相色谱-质谱:技术和统计考虑。","authors":"Sudhir Srivastava, Michael Merchant, Anil Rai, Shesh N Rai","doi":"10.35248/0974-276x.19.12.496","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>The quantitative measurements based on liquid chromatography (LC) coupled with mass spectrometry (MS) often suffer from the problem of missing values and data heterogeneity from technical variability. We considered a proteomics data set generated from human kidney biopsy material to investigate the technical effects of sample preparation and the quantitative MS.</p><p><strong>Methods: </strong>We studied the effect of tissue storage methods (TSMs) and tissue extraction methods (TEMs) on data analysis. There are two TSMs: frozen (FR) and FFPE (formalin-fixed paraffin embedded); and three TEMs: MAX, TX followed by MAX and SDS followed by MAX. We assessed the impact of different strategies to analyze the data while considering heterogeneity and MVs. We have used analysis of variance (ANOVA) model to study the effects due to various sources of variability.</p><p><strong>Results and conclusion: </strong>We found that the FFPE TSM is better than the FR TSM. We also found that the one-step TEM (MAX) is better than those of two-steps TEMs. Furthermore, we found the imputation method is a better approach than excluding the proteins with MVs or using unbalanced design.</p>","PeriodicalId":73911,"journal":{"name":"Journal of proteomics & bioinformatics","volume":"12 3","pages":"48-55"},"PeriodicalIF":0.0000,"publicationDate":"2019-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7059694/pdf/","citationCount":"13","resultStr":"{\"title\":\"Standardizing Proteomics Workflow for Liquid Chromatography-Mass Spectrometry: Technical and Statistical Considerations.\",\"authors\":\"Sudhir Srivastava, Michael Merchant, Anil Rai, Shesh N Rai\",\"doi\":\"10.35248/0974-276x.19.12.496\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>The quantitative measurements based on liquid chromatography (LC) coupled with mass spectrometry (MS) often suffer from the problem of missing values and data heterogeneity from technical variability. We considered a proteomics data set generated from human kidney biopsy material to investigate the technical effects of sample preparation and the quantitative MS.</p><p><strong>Methods: </strong>We studied the effect of tissue storage methods (TSMs) and tissue extraction methods (TEMs) on data analysis. There are two TSMs: frozen (FR) and FFPE (formalin-fixed paraffin embedded); and three TEMs: MAX, TX followed by MAX and SDS followed by MAX. We assessed the impact of different strategies to analyze the data while considering heterogeneity and MVs. We have used analysis of variance (ANOVA) model to study the effects due to various sources of variability.</p><p><strong>Results and conclusion: </strong>We found that the FFPE TSM is better than the FR TSM. We also found that the one-step TEM (MAX) is better than those of two-steps TEMs. Furthermore, we found the imputation method is a better approach than excluding the proteins with MVs or using unbalanced design.</p>\",\"PeriodicalId\":73911,\"journal\":{\"name\":\"Journal of proteomics & bioinformatics\",\"volume\":\"12 3\",\"pages\":\"48-55\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2019-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7059694/pdf/\",\"citationCount\":\"13\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of proteomics & bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.35248/0974-276x.19.12.496\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2019/4/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of proteomics & bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.35248/0974-276x.19.12.496","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/4/4 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Standardizing Proteomics Workflow for Liquid Chromatography-Mass Spectrometry: Technical and Statistical Considerations.
Introduction: The quantitative measurements based on liquid chromatography (LC) coupled with mass spectrometry (MS) often suffer from the problem of missing values and data heterogeneity from technical variability. We considered a proteomics data set generated from human kidney biopsy material to investigate the technical effects of sample preparation and the quantitative MS.
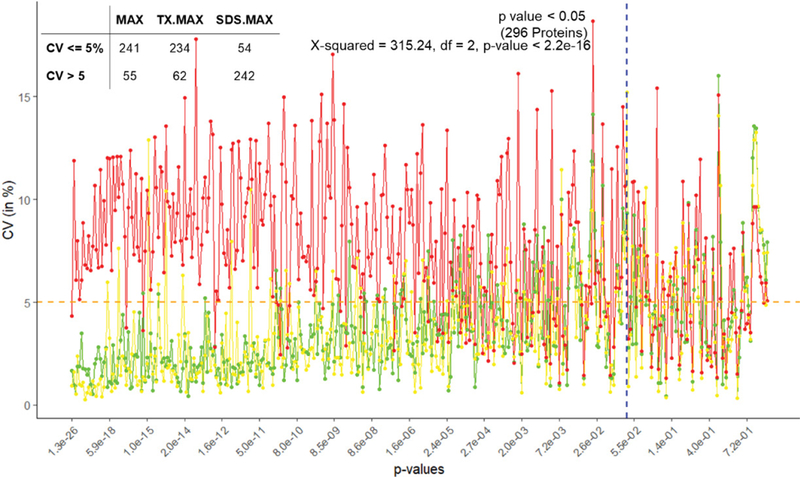
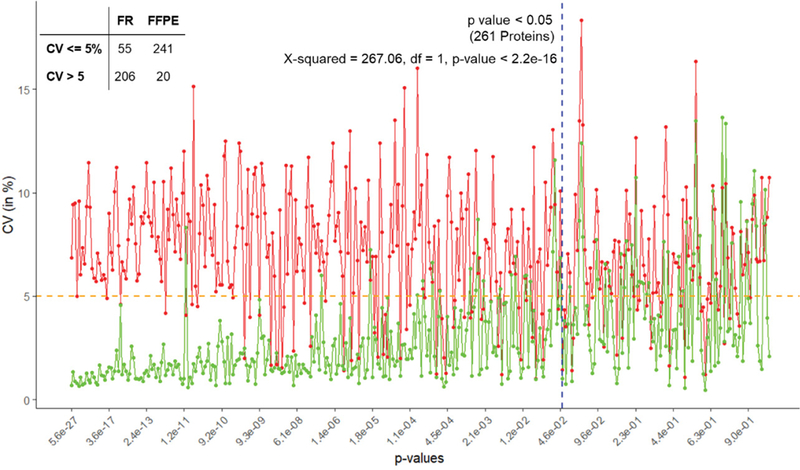
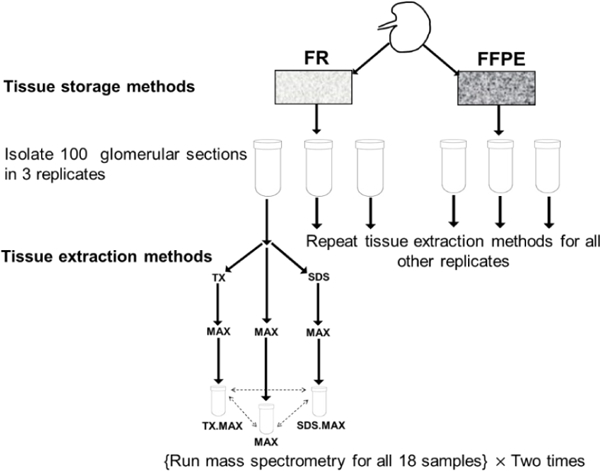
Methods: We studied the effect of tissue storage methods (TSMs) and tissue extraction methods (TEMs) on data analysis. There are two TSMs: frozen (FR) and FFPE (formalin-fixed paraffin embedded); and three TEMs: MAX, TX followed by MAX and SDS followed by MAX. We assessed the impact of different strategies to analyze the data while considering heterogeneity and MVs. We have used analysis of variance (ANOVA) model to study the effects due to various sources of variability.
Results and conclusion: We found that the FFPE TSM is better than the FR TSM. We also found that the one-step TEM (MAX) is better than those of two-steps TEMs. Furthermore, we found the imputation method is a better approach than excluding the proteins with MVs or using unbalanced design.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


