A Maver, G Čuturilo, Stojanović J Ruml, B Peterlin
{"title":"临床下一代测序揭示H3F3A基因是严重发育迟缓、智力残疾和生长迟缓相关的小头症的新潜在候选基因","authors":"A Maver, G Čuturilo, Stojanović J Ruml, B Peterlin","doi":"10.2478/bjmg-2019-0028","DOIUrl":null,"url":null,"abstract":"<p><p>Microcephaly is characterized by significant clinical and genetic heterogeneity, therefore reaching the genetic diagnosis remains challenging in this group of disorders. We describe a case of a girl with secondary microcephaly, associated with severe developmental delay, intellectual disability, growth retardation and dysmorphic features. For purposes of clinical genetic diagnostic testing, we performed trio whole exome sequencing in the proband and unaffected parents. We found a heterozygous <i>de novo</i> missense variant in the <i>H3F3A</i> gene in the proband (NM_ 002107.4: c.185T>G), which is absent from the gnomAD and from the Slovenian Genome databases. The identified variant affects a highly conserved leucine residue at position 62 of the histone H3 protein (H3.3) and is predicted to affect the physicochemical properties of the affected protein. Mouse models, which demonstrated involvement of H3.3 protein in the control of neuronal- and glial-specific gene expression patterns that control synaptic connectivity and behavioral plasticity. Additionally, we also identified similar cases reported in the ClinVar database. These arguments support the possible pathogenic role of the reported genetic variant and thus suggest a novel molecular mechanism for this syndromic form of microcephaly.</p>","PeriodicalId":520567,"journal":{"name":"Balkan journal of medical genetics : BJMG","volume":" ","pages":"65-68"},"PeriodicalIF":0.0000,"publicationDate":"2019-12-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/31/99/bjmg-22-065.PMC6956640.pdf","citationCount":"15","resultStr":"{\"title\":\"Clinical Next Generation Sequencing Reveals an <i>H3F3A</i> Gene as a New Potential Gene Candidate for Microcephaly Associated with Severe Developmental Delay, Intellectual Disability and Growth Retardation.\",\"authors\":\"A Maver, G Čuturilo, Stojanović J Ruml, B Peterlin\",\"doi\":\"10.2478/bjmg-2019-0028\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Microcephaly is characterized by significant clinical and genetic heterogeneity, therefore reaching the genetic diagnosis remains challenging in this group of disorders. We describe a case of a girl with secondary microcephaly, associated with severe developmental delay, intellectual disability, growth retardation and dysmorphic features. For purposes of clinical genetic diagnostic testing, we performed trio whole exome sequencing in the proband and unaffected parents. We found a heterozygous <i>de novo</i> missense variant in the <i>H3F3A</i> gene in the proband (NM_ 002107.4: c.185T>G), which is absent from the gnomAD and from the Slovenian Genome databases. The identified variant affects a highly conserved leucine residue at position 62 of the histone H3 protein (H3.3) and is predicted to affect the physicochemical properties of the affected protein. Mouse models, which demonstrated involvement of H3.3 protein in the control of neuronal- and glial-specific gene expression patterns that control synaptic connectivity and behavioral plasticity. Additionally, we also identified similar cases reported in the ClinVar database. These arguments support the possible pathogenic role of the reported genetic variant and thus suggest a novel molecular mechanism for this syndromic form of microcephaly.</p>\",\"PeriodicalId\":520567,\"journal\":{\"name\":\"Balkan journal of medical genetics : BJMG\",\"volume\":\" \",\"pages\":\"65-68\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2019-12-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/31/99/bjmg-22-065.PMC6956640.pdf\",\"citationCount\":\"15\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Balkan journal of medical genetics : BJMG\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.2478/bjmg-2019-0028\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2019/12/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Balkan journal of medical genetics : BJMG","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2478/bjmg-2019-0028","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/12/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Clinical Next Generation Sequencing Reveals an H3F3A Gene as a New Potential Gene Candidate for Microcephaly Associated with Severe Developmental Delay, Intellectual Disability and Growth Retardation.
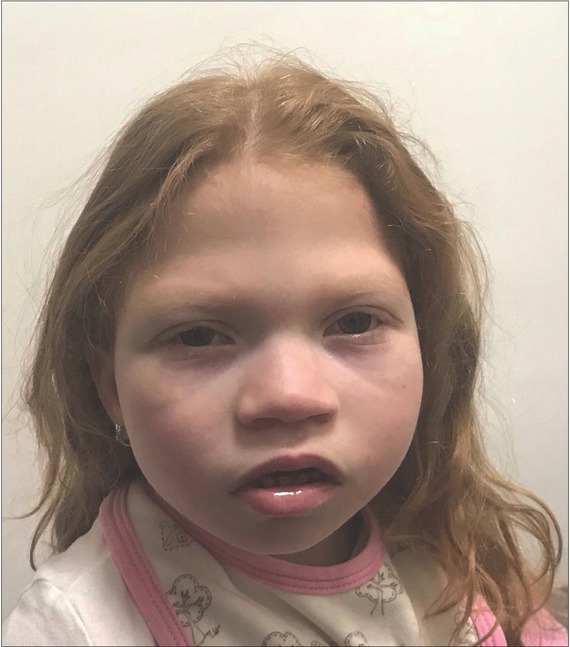
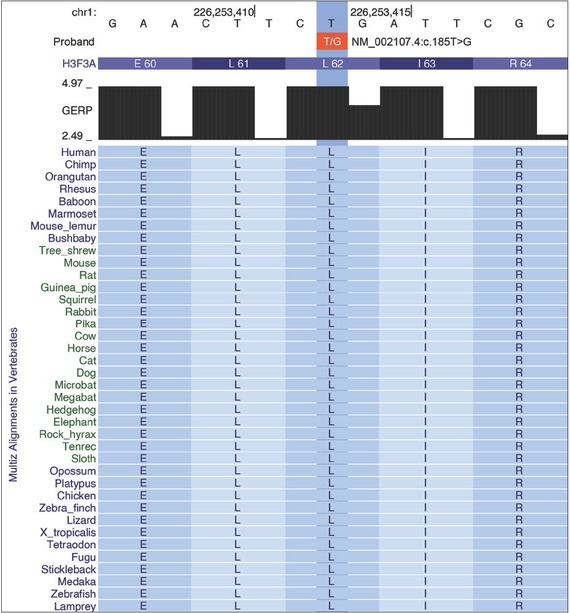
Microcephaly is characterized by significant clinical and genetic heterogeneity, therefore reaching the genetic diagnosis remains challenging in this group of disorders. We describe a case of a girl with secondary microcephaly, associated with severe developmental delay, intellectual disability, growth retardation and dysmorphic features. For purposes of clinical genetic diagnostic testing, we performed trio whole exome sequencing in the proband and unaffected parents. We found a heterozygous de novo missense variant in the H3F3A gene in the proband (NM_ 002107.4: c.185T>G), which is absent from the gnomAD and from the Slovenian Genome databases. The identified variant affects a highly conserved leucine residue at position 62 of the histone H3 protein (H3.3) and is predicted to affect the physicochemical properties of the affected protein. Mouse models, which demonstrated involvement of H3.3 protein in the control of neuronal- and glial-specific gene expression patterns that control synaptic connectivity and behavioral plasticity. Additionally, we also identified similar cases reported in the ClinVar database. These arguments support the possible pathogenic role of the reported genetic variant and thus suggest a novel molecular mechanism for this syndromic form of microcephaly.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


