Kridsadakorn Chaichoompu, Fentaw Abegaz, Sissades Tongsima, Philip James Shaw, Anavaj Sakuntabhai, Luísa Pereira, Kristel Van Steen
{"title":"IPCAPS:一个R包迭代修剪捕捉人口结构。","authors":"Kridsadakorn Chaichoompu, Fentaw Abegaz, Sissades Tongsima, Philip James Shaw, Anavaj Sakuntabhai, Luísa Pereira, Kristel Van Steen","doi":"10.1186/s13029-019-0072-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Resolving population genetic structure is challenging, especially when dealing with closely related or geographically confined populations. Although Principal Component Analysis (PCA)-based methods and genomic variation with single nucleotide polymorphisms (SNPs) are widely used to describe shared genetic ancestry, improvements can be made especially when fine-scale population structure is the target.</p><p><strong>Results: </strong>This work presents an R package called IPCAPS, which uses SNP information for resolving possibly fine-scale population structure. The IPCAPS routines are built on the iterative pruning Principal Component Analysis (ipPCA) framework that systematically assigns individuals to genetically similar subgroups. In each iteration, our tool is able to detect and eliminate outliers, hereby avoiding severe misclassification errors.</p><p><strong>Conclusions: </strong>IPCAPS supports different measurement scales for variables used to identify substructure. Hence, panels of gene expression and methylation data can be accommodated as well. The tool can also be applied in patient sub-phenotyping contexts. IPCAPS is developed in R and is freely available from http://bio3.giga.ulg.ac.be/ipcaps.</p>","PeriodicalId":35052,"journal":{"name":"Source Code for Biology and Medicine","volume":"14 ","pages":"2"},"PeriodicalIF":0.0000,"publicationDate":"2019-03-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13029-019-0072-6","citationCount":"13","resultStr":"{\"title\":\"IPCAPS: an R package for iterative pruning to capture population structure.\",\"authors\":\"Kridsadakorn Chaichoompu, Fentaw Abegaz, Sissades Tongsima, Philip James Shaw, Anavaj Sakuntabhai, Luísa Pereira, Kristel Van Steen\",\"doi\":\"10.1186/s13029-019-0072-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Resolving population genetic structure is challenging, especially when dealing with closely related or geographically confined populations. Although Principal Component Analysis (PCA)-based methods and genomic variation with single nucleotide polymorphisms (SNPs) are widely used to describe shared genetic ancestry, improvements can be made especially when fine-scale population structure is the target.</p><p><strong>Results: </strong>This work presents an R package called IPCAPS, which uses SNP information for resolving possibly fine-scale population structure. The IPCAPS routines are built on the iterative pruning Principal Component Analysis (ipPCA) framework that systematically assigns individuals to genetically similar subgroups. In each iteration, our tool is able to detect and eliminate outliers, hereby avoiding severe misclassification errors.</p><p><strong>Conclusions: </strong>IPCAPS supports different measurement scales for variables used to identify substructure. Hence, panels of gene expression and methylation data can be accommodated as well. The tool can also be applied in patient sub-phenotyping contexts. IPCAPS is developed in R and is freely available from http://bio3.giga.ulg.ac.be/ipcaps.</p>\",\"PeriodicalId\":35052,\"journal\":{\"name\":\"Source Code for Biology and Medicine\",\"volume\":\"14 \",\"pages\":\"2\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2019-03-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13029-019-0072-6\",\"citationCount\":\"13\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Source Code for Biology and Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13029-019-0072-6\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2019/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"Decision Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Source Code for Biology and Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13029-019-0072-6","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Decision Sciences","Score":null,"Total":0}
IPCAPS: an R package for iterative pruning to capture population structure.
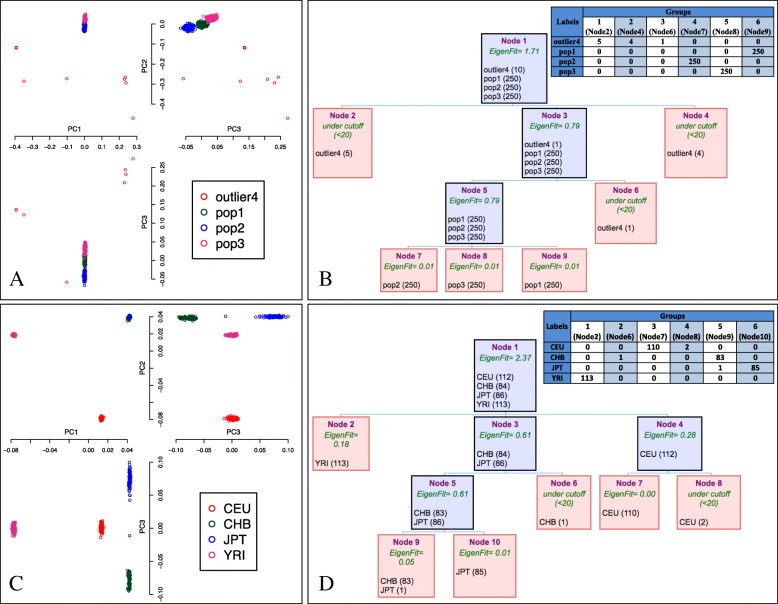
Background: Resolving population genetic structure is challenging, especially when dealing with closely related or geographically confined populations. Although Principal Component Analysis (PCA)-based methods and genomic variation with single nucleotide polymorphisms (SNPs) are widely used to describe shared genetic ancestry, improvements can be made especially when fine-scale population structure is the target.
Results: This work presents an R package called IPCAPS, which uses SNP information for resolving possibly fine-scale population structure. The IPCAPS routines are built on the iterative pruning Principal Component Analysis (ipPCA) framework that systematically assigns individuals to genetically similar subgroups. In each iteration, our tool is able to detect and eliminate outliers, hereby avoiding severe misclassification errors.
Conclusions: IPCAPS supports different measurement scales for variables used to identify substructure. Hence, panels of gene expression and methylation data can be accommodated as well. The tool can also be applied in patient sub-phenotyping contexts. IPCAPS is developed in R and is freely available from http://bio3.giga.ulg.ac.be/ipcaps.
期刊介绍:
Source Code for Biology and Medicine is a peer-reviewed open access, online journal that publishes articles on source code employed over a wide range of applications in biology and medicine. The journal"s aim is to publish source code for distribution and use in the public domain in order to advance biological and medical research. Through this dissemination, it may be possible to shorten the time required for solving certain computational problems for which there is limited source code availability or resources.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


