{"title":"qPCR数据分析:iconoclasm效果更好","authors":"Joel Tellinghuisen , Andrej-Nikolai Spiess","doi":"10.1016/j.bdq.2019.100084","DOIUrl":null,"url":null,"abstract":"<div><p>The standard approach for quantitative estimation of genetic materials with qPCR is calibration with known concentrations for the target substance, in which estimates of the quantification cycle (<em>C<sub>q</sub></em>) are fitted to a straight-line function of log(<em>N</em><sub>0</sub>), where <em>N</em><sub>0</sub> is the initial number of target molecules. The location of <em>C<sub>q</sub></em> for the unknown on this line then yields its <em>N</em><sub>0</sub>. The most widely used definition for <em>C<sub>q</sub></em> is an absolute threshold that falls in the early growth cycles. This usage is flawed as commonly implemented: threshold set very close to the baseline level, which is estimated separately, from designated \"baseline cycles.\" The absolute threshold is especially poor for dealing with the scale variability often observed for growth profiles. Scale-independent markers, like the first derivative maximum (FDM) and a relative threshold (<em>C<sub>r</sub></em>) avoid this problem. We describe improved methods for estimating these and other <em>C<sub>q</sub></em> markers and their standard errors, from a nonlinear algorithm that fits growth profiles to a 4-parameter log-logistic function plus a baseline function. By examining six multidilution, multireplicate qPCR data sets, we find that nonlinear expressions are often preferred statistically for the dependence of <em>C<sub>q</sub></em> on log(<em>N</em><sub>0</sub>). This means that the amplification efficiency <em>E</em> depends on <em>N</em><sub>0</sub>, in violation of another tenet of qPCR analysis. Neglect of calibration nonlinearity leads to biased estimates of the unknown. By logic, <em>E</em> estimates from calibration fitting pertain to the earliest baseline cycles, <em>not</em> the early growth cycles used to estimate <em>E</em> from growth profiles for single reactions. This raises concern about the use of the latter in lengthy extrapolations to estimate <em>N</em><sub>0</sub>. Finally, we observe that replicate ensemble standard deviations greatly exceed predictions, implying that much better results can be achieved from qPCR through better experimental procedures, which likely include reducing pipette volume uncertainty.</p></div>","PeriodicalId":38073,"journal":{"name":"Biomolecular Detection and Quantification","volume":"17 ","pages":"Article 100084"},"PeriodicalIF":0.0000,"publicationDate":"2019-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.bdq.2019.100084","citationCount":"9","resultStr":"{\"title\":\"qPCR data analysis: Better results through iconoclasm\",\"authors\":\"Joel Tellinghuisen , Andrej-Nikolai Spiess\",\"doi\":\"10.1016/j.bdq.2019.100084\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The standard approach for quantitative estimation of genetic materials with qPCR is calibration with known concentrations for the target substance, in which estimates of the quantification cycle (<em>C<sub>q</sub></em>) are fitted to a straight-line function of log(<em>N</em><sub>0</sub>), where <em>N</em><sub>0</sub> is the initial number of target molecules. The location of <em>C<sub>q</sub></em> for the unknown on this line then yields its <em>N</em><sub>0</sub>. The most widely used definition for <em>C<sub>q</sub></em> is an absolute threshold that falls in the early growth cycles. This usage is flawed as commonly implemented: threshold set very close to the baseline level, which is estimated separately, from designated \\\"baseline cycles.\\\" The absolute threshold is especially poor for dealing with the scale variability often observed for growth profiles. Scale-independent markers, like the first derivative maximum (FDM) and a relative threshold (<em>C<sub>r</sub></em>) avoid this problem. We describe improved methods for estimating these and other <em>C<sub>q</sub></em> markers and their standard errors, from a nonlinear algorithm that fits growth profiles to a 4-parameter log-logistic function plus a baseline function. By examining six multidilution, multireplicate qPCR data sets, we find that nonlinear expressions are often preferred statistically for the dependence of <em>C<sub>q</sub></em> on log(<em>N</em><sub>0</sub>). This means that the amplification efficiency <em>E</em> depends on <em>N</em><sub>0</sub>, in violation of another tenet of qPCR analysis. Neglect of calibration nonlinearity leads to biased estimates of the unknown. By logic, <em>E</em> estimates from calibration fitting pertain to the earliest baseline cycles, <em>not</em> the early growth cycles used to estimate <em>E</em> from growth profiles for single reactions. This raises concern about the use of the latter in lengthy extrapolations to estimate <em>N</em><sub>0</sub>. Finally, we observe that replicate ensemble standard deviations greatly exceed predictions, implying that much better results can be achieved from qPCR through better experimental procedures, which likely include reducing pipette volume uncertainty.</p></div>\",\"PeriodicalId\":38073,\"journal\":{\"name\":\"Biomolecular Detection and Quantification\",\"volume\":\"17 \",\"pages\":\"Article 100084\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2019-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1016/j.bdq.2019.100084\",\"citationCount\":\"9\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biomolecular Detection and Quantification\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S221475351830010X\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biomolecular Detection and Quantification","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S221475351830010X","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
qPCR data analysis: Better results through iconoclasm
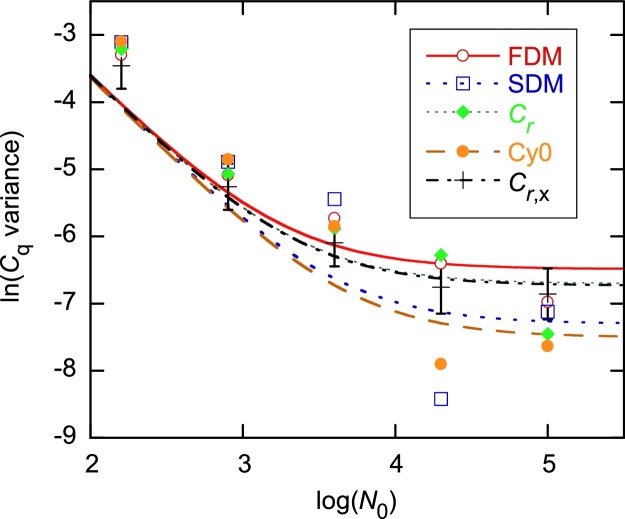
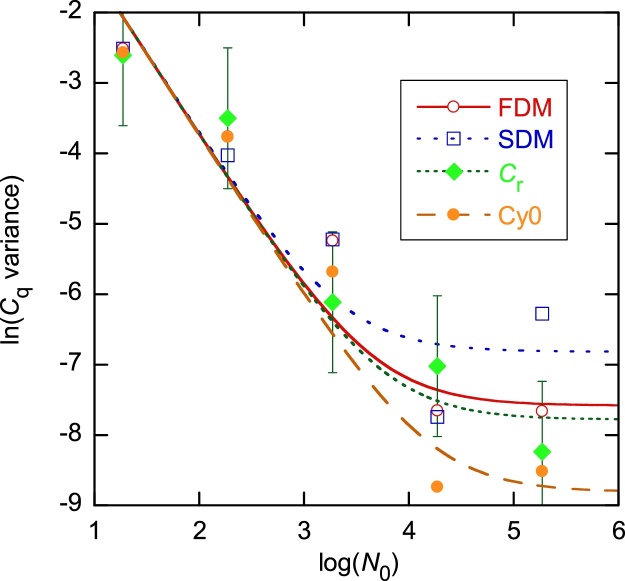
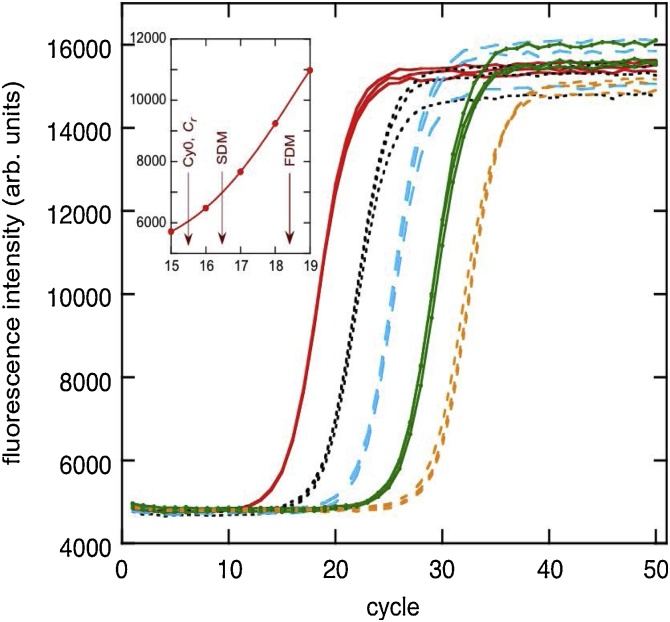
The standard approach for quantitative estimation of genetic materials with qPCR is calibration with known concentrations for the target substance, in which estimates of the quantification cycle (Cq) are fitted to a straight-line function of log(N0), where N0 is the initial number of target molecules. The location of Cq for the unknown on this line then yields its N0. The most widely used definition for Cq is an absolute threshold that falls in the early growth cycles. This usage is flawed as commonly implemented: threshold set very close to the baseline level, which is estimated separately, from designated "baseline cycles." The absolute threshold is especially poor for dealing with the scale variability often observed for growth profiles. Scale-independent markers, like the first derivative maximum (FDM) and a relative threshold (Cr) avoid this problem. We describe improved methods for estimating these and other Cq markers and their standard errors, from a nonlinear algorithm that fits growth profiles to a 4-parameter log-logistic function plus a baseline function. By examining six multidilution, multireplicate qPCR data sets, we find that nonlinear expressions are often preferred statistically for the dependence of Cq on log(N0). This means that the amplification efficiency E depends on N0, in violation of another tenet of qPCR analysis. Neglect of calibration nonlinearity leads to biased estimates of the unknown. By logic, E estimates from calibration fitting pertain to the earliest baseline cycles, not the early growth cycles used to estimate E from growth profiles for single reactions. This raises concern about the use of the latter in lengthy extrapolations to estimate N0. Finally, we observe that replicate ensemble standard deviations greatly exceed predictions, implying that much better results can be achieved from qPCR through better experimental procedures, which likely include reducing pipette volume uncertainty.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


