结合蛋白质组学和转录组学数据的转录因子的蛋白质- dna相互作用预测
Q4 Biochemistry, Genetics and Molecular Biology
引用次数: 4
摘要
我们比较了基于位置权矩阵的转录因子(TF)结合位点预测方法,使用ChIP-seq数据的选定部分,并借助部分AUC测量(限于假阳性率0.1,这是与TF搜索在基因组尺度上的应用最相关的)。通过对三种预测方法——相加法、乘法法和基于信息向量法(information-vector based, MATCH)的比较,发现MATCH方法对大多数被测转录因子具有优势。我们证明了TF位点鉴定方法的应用可以帮助将信号网络的蛋白质组学和磷酸化蛋白质组学世界与基因调控和转录组学世界联系起来。本文章由计算机程序翻译,如有差异,请以英文原文为准。
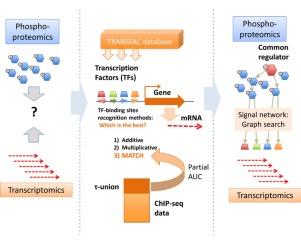
Prediction of protein-DNA interactions of transcription factors linking proteomics and transcriptomics data
We compared positional weight matrix-based prediction methods for transcription factor (TF) binding sites using selected fraction of ChIP-seq data with the help of partial AUC measure (limited to false positive rate 0.1, that is the most relevant for the application of the TF search in the genome scale). Comparison of three prediction methods—additive, multiplicative and information-vector based (MATCH) showed an advantage of the MATCH method for majority of transcription factors tested. We demonstrated that application of TF site identifying methods can help to connect the proteomics and phosphoproteomics world of signaling networks to gene regulation and transcriptomics world.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

EuPA Open Proteomics
Biochemistry, Genetics and Molecular Biology-Biochemistry
自引率
0.00%
发文量
0
审稿时长
103 days
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


