Krzysztof Marciniec, Rafał Kurczab, Maria Książek, Ewa Bębenek, Elwira Chrobak, Grzegorz Satała, Andrzej J Bojarski, Joachim Kusz, Paweł Zajdel
{"title":"影响卤素键的结构决定因素:阿立哌唑azinesulfonamide类似物作为5-HT1A、5-HT7和D2受体配体的案例研究","authors":"Krzysztof Marciniec, Rafał Kurczab, Maria Książek, Ewa Bębenek, Elwira Chrobak, Grzegorz Satała, Andrzej J Bojarski, Joachim Kusz, Paweł Zajdel","doi":"10.1186/s13065-018-0422-5","DOIUrl":null,"url":null,"abstract":"<p><p>A series of azinesulfonamide derivatives of long-chain arylpiperazines with variable-length alkylene spacers between sulfonamide and 4-arylpiperazine moiety is designed, synthesized, and biologically evaluated. In vitro methods are used to determine their affinity for serotonin 5-HT<sub>1A</sub>, 5-HT<sub>6</sub>, 5-HT<sub>7</sub>, and dopamine D<sub>2</sub> receptors. X-ray analysis, two-dimensional NMR conformational studies, and docking into the 5-HT<sub>1A</sub> and 5-HT<sub>7</sub> receptor models are then conducted to investigate the conformational preferences of selected serotonin receptor ligands in different environments. The bent conformation of tetramethylene derivatives is found in a solid state, in dimethyl sulfoxide, and as a global energy minimum during conformational analysis in a simulated water environment. Furthermore, ligand geometry in top-scored complexes is also bent, with one torsion angle in the spacer (τ<sub>2</sub>) in synclinal conformation. Molecular docking studies indicate the role of halogen bonding in complexes of the most potent ligands and target receptors.</p>","PeriodicalId":9842,"journal":{"name":"Chemistry Central Journal","volume":"12 1","pages":"55"},"PeriodicalIF":0.0000,"publicationDate":"2018-05-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13065-018-0422-5","citationCount":"6","resultStr":"{\"title\":\"Structural determinants influencing halogen bonding: a case study on azinesulfonamide analogs of aripiprazole as 5-HT<sub>1A</sub>, 5-HT<sub>7</sub>, and D<sub>2</sub> receptor ligands.\",\"authors\":\"Krzysztof Marciniec, Rafał Kurczab, Maria Książek, Ewa Bębenek, Elwira Chrobak, Grzegorz Satała, Andrzej J Bojarski, Joachim Kusz, Paweł Zajdel\",\"doi\":\"10.1186/s13065-018-0422-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>A series of azinesulfonamide derivatives of long-chain arylpiperazines with variable-length alkylene spacers between sulfonamide and 4-arylpiperazine moiety is designed, synthesized, and biologically evaluated. In vitro methods are used to determine their affinity for serotonin 5-HT<sub>1A</sub>, 5-HT<sub>6</sub>, 5-HT<sub>7</sub>, and dopamine D<sub>2</sub> receptors. X-ray analysis, two-dimensional NMR conformational studies, and docking into the 5-HT<sub>1A</sub> and 5-HT<sub>7</sub> receptor models are then conducted to investigate the conformational preferences of selected serotonin receptor ligands in different environments. The bent conformation of tetramethylene derivatives is found in a solid state, in dimethyl sulfoxide, and as a global energy minimum during conformational analysis in a simulated water environment. Furthermore, ligand geometry in top-scored complexes is also bent, with one torsion angle in the spacer (τ<sub>2</sub>) in synclinal conformation. Molecular docking studies indicate the role of halogen bonding in complexes of the most potent ligands and target receptors.</p>\",\"PeriodicalId\":9842,\"journal\":{\"name\":\"Chemistry Central Journal\",\"volume\":\"12 1\",\"pages\":\"55\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2018-05-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13065-018-0422-5\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chemistry Central Journal\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13065-018-0422-5\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Chemistry\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemistry Central Journal","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13065-018-0422-5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Chemistry","Score":null,"Total":0}
Structural determinants influencing halogen bonding: a case study on azinesulfonamide analogs of aripiprazole as 5-HT1A, 5-HT7, and D2 receptor ligands.
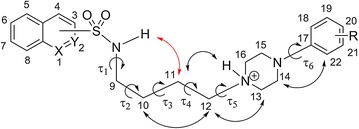
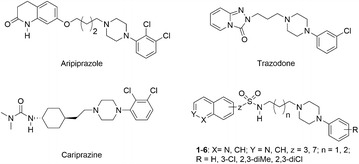
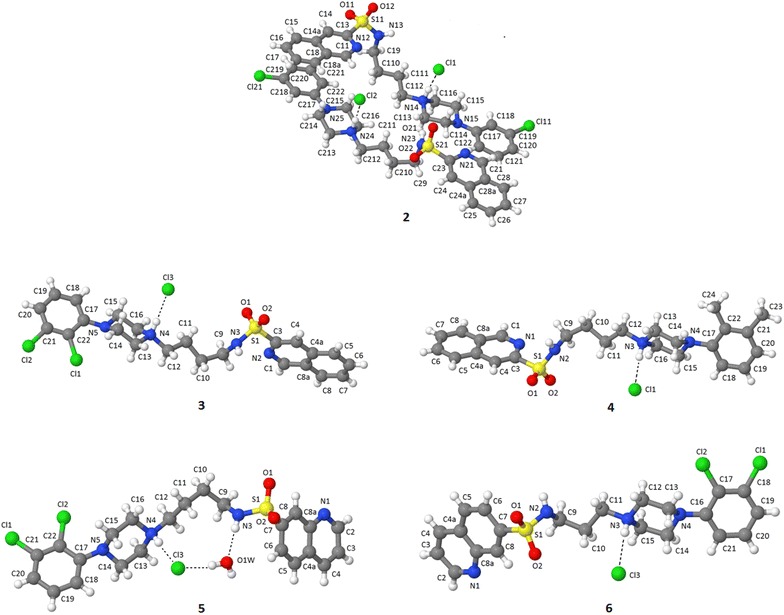
A series of azinesulfonamide derivatives of long-chain arylpiperazines with variable-length alkylene spacers between sulfonamide and 4-arylpiperazine moiety is designed, synthesized, and biologically evaluated. In vitro methods are used to determine their affinity for serotonin 5-HT1A, 5-HT6, 5-HT7, and dopamine D2 receptors. X-ray analysis, two-dimensional NMR conformational studies, and docking into the 5-HT1A and 5-HT7 receptor models are then conducted to investigate the conformational preferences of selected serotonin receptor ligands in different environments. The bent conformation of tetramethylene derivatives is found in a solid state, in dimethyl sulfoxide, and as a global energy minimum during conformational analysis in a simulated water environment. Furthermore, ligand geometry in top-scored complexes is also bent, with one torsion angle in the spacer (τ2) in synclinal conformation. Molecular docking studies indicate the role of halogen bonding in complexes of the most potent ligands and target receptors.
期刊介绍:
BMC Chemistry is an open access, peer reviewed journal that considers all articles in the broad field of chemistry, including research on fundamental concepts, new developments and the application of chemical sciences to broad range of research fields, industry, and other disciplines. It provides an inclusive platform for the dissemination and discussion of chemistry to aid the advancement of all areas of research.
Sections:
-Analytical Chemistry
-Organic Chemistry
-Environmental and Energy Chemistry
-Agricultural and Food Chemistry
-Inorganic Chemistry
-Medicinal Chemistry
-Physical Chemistry
-Materials and Macromolecular Chemistry
-Green and Sustainable Chemistry

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


