{"title":"FSH:利用相邻哈希的快速间隔种子哈希。","authors":"Samuele Girotto, Matteo Comin, Cinzia Pizzi","doi":"10.1186/s13015-018-0125-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Patterns with wildcards in specified positions, namely <i>spaced seeds</i>, are increasingly used instead of <i>k</i>-mers in many bioinformatics applications that require indexing, querying and rapid similarity search, as they can provide better sensitivity. Many of these applications require to compute the hashing of each position in the input sequences with respect to the given spaced seed, or to multiple spaced seeds. While the hashing of <i>k</i>-mers can be rapidly computed by exploiting the large overlap between consecutive <i>k</i>-mers, spaced seeds hashing is usually computed from scratch for each position in the input sequence, thus resulting in slower processing.</p><p><strong>Results: </strong>The method proposed in this paper, fast spaced-seed hashing (FSH), exploits the similarity of the hash values of spaced seeds computed at adjacent positions in the input sequence. In our experiments we compute the hash for each positions of metagenomics reads from several datasets, with respect to different spaced seeds. We also propose a generalized version of the algorithm for the simultaneous computation of multiple spaced seeds hashing. In the experiments, our algorithm can compute the hashing values of spaced seeds with a speedup, with respect to the traditional approach, between 1.6[Formula: see text] to 5.3[Formula: see text], depending on the structure of the spaced seed.</p><p><strong>Conclusions: </strong>Spaced seed hashing is a routine task for several bioinformatics application. FSH allows to perform this task efficiently and raise the question of whether other hashing can be exploited to further improve the speed up. This has the potential of major impact in the field, making spaced seed applications not only accurate, but also faster and more efficient.</p><p><strong>Availability: </strong>The software FSH is freely available for academic use at: https://bitbucket.org/samu661/fsh/overview.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"13 ","pages":"8"},"PeriodicalIF":1.5000,"publicationDate":"2018-03-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13015-018-0125-4","citationCount":"7","resultStr":"{\"title\":\"FSH: fast spaced seed hashing exploiting adjacent hashes.\",\"authors\":\"Samuele Girotto, Matteo Comin, Cinzia Pizzi\",\"doi\":\"10.1186/s13015-018-0125-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Patterns with wildcards in specified positions, namely <i>spaced seeds</i>, are increasingly used instead of <i>k</i>-mers in many bioinformatics applications that require indexing, querying and rapid similarity search, as they can provide better sensitivity. Many of these applications require to compute the hashing of each position in the input sequences with respect to the given spaced seed, or to multiple spaced seeds. While the hashing of <i>k</i>-mers can be rapidly computed by exploiting the large overlap between consecutive <i>k</i>-mers, spaced seeds hashing is usually computed from scratch for each position in the input sequence, thus resulting in slower processing.</p><p><strong>Results: </strong>The method proposed in this paper, fast spaced-seed hashing (FSH), exploits the similarity of the hash values of spaced seeds computed at adjacent positions in the input sequence. In our experiments we compute the hash for each positions of metagenomics reads from several datasets, with respect to different spaced seeds. We also propose a generalized version of the algorithm for the simultaneous computation of multiple spaced seeds hashing. In the experiments, our algorithm can compute the hashing values of spaced seeds with a speedup, with respect to the traditional approach, between 1.6[Formula: see text] to 5.3[Formula: see text], depending on the structure of the spaced seed.</p><p><strong>Conclusions: </strong>Spaced seed hashing is a routine task for several bioinformatics application. FSH allows to perform this task efficiently and raise the question of whether other hashing can be exploited to further improve the speed up. This has the potential of major impact in the field, making spaced seed applications not only accurate, but also faster and more efficient.</p><p><strong>Availability: </strong>The software FSH is freely available for academic use at: https://bitbucket.org/samu661/fsh/overview.</p>\",\"PeriodicalId\":50823,\"journal\":{\"name\":\"Algorithms for Molecular Biology\",\"volume\":\"13 \",\"pages\":\"8\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2018-03-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13015-018-0125-4\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Algorithms for Molecular Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13015-018-0125-4\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2018/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-018-0125-4","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
FSH: fast spaced seed hashing exploiting adjacent hashes.
Background: Patterns with wildcards in specified positions, namely spaced seeds, are increasingly used instead of k-mers in many bioinformatics applications that require indexing, querying and rapid similarity search, as they can provide better sensitivity. Many of these applications require to compute the hashing of each position in the input sequences with respect to the given spaced seed, or to multiple spaced seeds. While the hashing of k-mers can be rapidly computed by exploiting the large overlap between consecutive k-mers, spaced seeds hashing is usually computed from scratch for each position in the input sequence, thus resulting in slower processing.
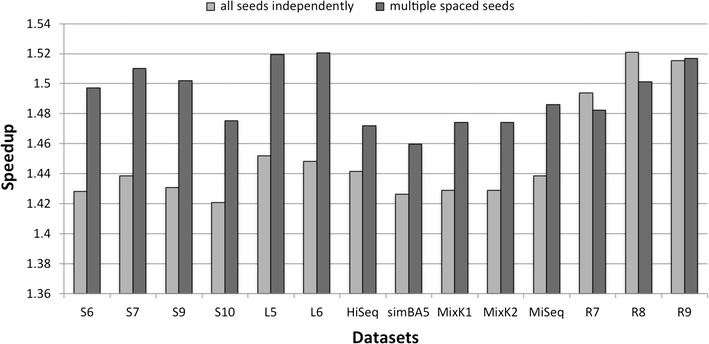
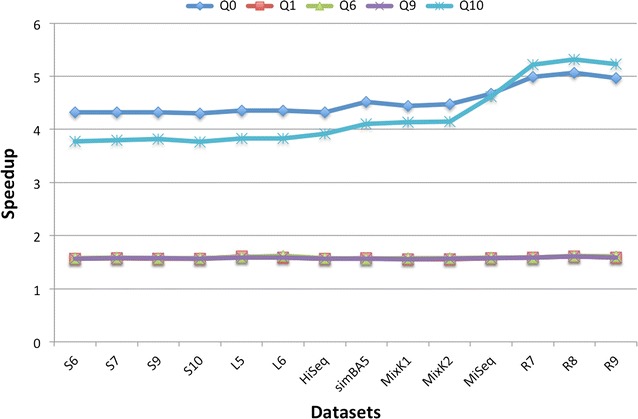
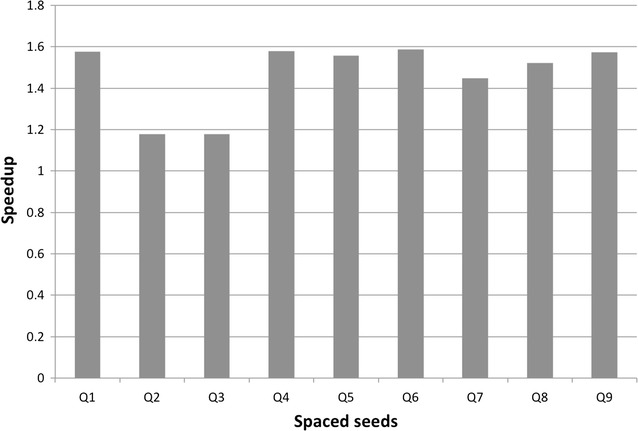
Results: The method proposed in this paper, fast spaced-seed hashing (FSH), exploits the similarity of the hash values of spaced seeds computed at adjacent positions in the input sequence. In our experiments we compute the hash for each positions of metagenomics reads from several datasets, with respect to different spaced seeds. We also propose a generalized version of the algorithm for the simultaneous computation of multiple spaced seeds hashing. In the experiments, our algorithm can compute the hashing values of spaced seeds with a speedup, with respect to the traditional approach, between 1.6[Formula: see text] to 5.3[Formula: see text], depending on the structure of the spaced seed.
Conclusions: Spaced seed hashing is a routine task for several bioinformatics application. FSH allows to perform this task efficiently and raise the question of whether other hashing can be exploited to further improve the speed up. This has the potential of major impact in the field, making spaced seed applications not only accurate, but also faster and more efficient.
Availability: The software FSH is freely available for academic use at: https://bitbucket.org/samu661/fsh/overview.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


