Karen M Rothacker, Katie L Ayers, Dave Tang, Kiranjit Joshi, Jocelyn A van den Bergen, Gorjana Robevska, Naeem Samnakay, Lakshmi Nagarajan, Kate Francis, Andrew H Sinclair, Catherine S Choong
{"title":"一种新的,纯合突变的沙漠刺猬(DHH)在46,XY患者的睾丸发育不良,表现为原发性闭经:一个病例报告。","authors":"Karen M Rothacker, Katie L Ayers, Dave Tang, Kiranjit Joshi, Jocelyn A van den Bergen, Gorjana Robevska, Naeem Samnakay, Lakshmi Nagarajan, Kate Francis, Andrew H Sinclair, Catherine S Choong","doi":"10.1186/s13633-018-0056-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong><i>Desert hedgehog</i> (<i>DHH</i>) mutations have been described in only a limited number of individuals with 46, XY disorders of sex development (DSD) presenting as either partial or complete gonadal dysgenesis. Gonadal tumours and peripheral neuropathy have been associated with <i>DHH</i> mutations. Herein we report a novel, homozygous mutation of <i>DHH</i> identified through a targeted, massively parallel sequencing (MPS) DSD panel, in a patient presenting with partial gonadal dysgenesis. This novel mutation is two amino acids away from a previously described mutation in a patient who presented with complete gonadal dysgenesis. Adding to the complexity of work-up, our patient also expressed gender identity concern.</p><p><strong>Case presentation: </strong>A 14-year-old, phenotypic female presented with primary amenorrhoea and absent secondary sex characteristics. Investigations revealed elevated gonadotrophins with low oestradiol, testosterone of 0.6 nmol/L and a 46, XY karyotype. Müllerian structures were not seen on pelvic ultrasound or laparoscopically and gonadal biopsies demonstrated dysgenetic testes without neoplasia (partial gonadal dysgenesis). The patient expressed gender identity confusion upon initial notification of investigation findings. Formal psychiatric evaluation excluded gender dysphoria. Genetic analysis was performed using a targeted, MPS DSD panel of 64 diagnostic and 927 research candidate genes. This identified a novel, homozygous mutation in exon 2 of <i>DHH</i> (DHH:NM_021044:exon2:c.G491C:p.R164P). With this finding our patient was screened for the possibility of peripheral neuropathy which was not evident clinically nor on investigation. She was commenced on oestrogen for pubertal induction.</p><p><strong>Conclusion: </strong>The evaluation of patients with DSD is associated with considerable psychological distress. Targeted MPS enables an affordable and efficient method for diagnosis of 46, XY DSD cases. Identifying a genetic diagnosis may inform clinical management and in this case directed screening for peripheral neuropathy. In addition to the structural location of the mutation other interacting factors may influence phenotypic expression in homozygous <i>DHH</i> mutations.</p>","PeriodicalId":14271,"journal":{"name":"International Journal of Pediatric Endocrinology","volume":"2018 ","pages":"2"},"PeriodicalIF":0.0000,"publicationDate":"2018-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13633-018-0056-3","citationCount":"13","resultStr":"{\"title\":\"A novel, homozygous mutation in <i>desert hedgehog</i> (<i>DHH</i>) in a 46, XY patient with dysgenetic testes presenting with primary amenorrhoea: a case report.\",\"authors\":\"Karen M Rothacker, Katie L Ayers, Dave Tang, Kiranjit Joshi, Jocelyn A van den Bergen, Gorjana Robevska, Naeem Samnakay, Lakshmi Nagarajan, Kate Francis, Andrew H Sinclair, Catherine S Choong\",\"doi\":\"10.1186/s13633-018-0056-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong><i>Desert hedgehog</i> (<i>DHH</i>) mutations have been described in only a limited number of individuals with 46, XY disorders of sex development (DSD) presenting as either partial or complete gonadal dysgenesis. Gonadal tumours and peripheral neuropathy have been associated with <i>DHH</i> mutations. Herein we report a novel, homozygous mutation of <i>DHH</i> identified through a targeted, massively parallel sequencing (MPS) DSD panel, in a patient presenting with partial gonadal dysgenesis. This novel mutation is two amino acids away from a previously described mutation in a patient who presented with complete gonadal dysgenesis. Adding to the complexity of work-up, our patient also expressed gender identity concern.</p><p><strong>Case presentation: </strong>A 14-year-old, phenotypic female presented with primary amenorrhoea and absent secondary sex characteristics. Investigations revealed elevated gonadotrophins with low oestradiol, testosterone of 0.6 nmol/L and a 46, XY karyotype. Müllerian structures were not seen on pelvic ultrasound or laparoscopically and gonadal biopsies demonstrated dysgenetic testes without neoplasia (partial gonadal dysgenesis). The patient expressed gender identity confusion upon initial notification of investigation findings. Formal psychiatric evaluation excluded gender dysphoria. Genetic analysis was performed using a targeted, MPS DSD panel of 64 diagnostic and 927 research candidate genes. This identified a novel, homozygous mutation in exon 2 of <i>DHH</i> (DHH:NM_021044:exon2:c.G491C:p.R164P). With this finding our patient was screened for the possibility of peripheral neuropathy which was not evident clinically nor on investigation. She was commenced on oestrogen for pubertal induction.</p><p><strong>Conclusion: </strong>The evaluation of patients with DSD is associated with considerable psychological distress. Targeted MPS enables an affordable and efficient method for diagnosis of 46, XY DSD cases. Identifying a genetic diagnosis may inform clinical management and in this case directed screening for peripheral neuropathy. In addition to the structural location of the mutation other interacting factors may influence phenotypic expression in homozygous <i>DHH</i> mutations.</p>\",\"PeriodicalId\":14271,\"journal\":{\"name\":\"International Journal of Pediatric Endocrinology\",\"volume\":\"2018 \",\"pages\":\"2\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2018-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13633-018-0056-3\",\"citationCount\":\"13\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Pediatric Endocrinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13633-018-0056-3\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2018/3/2 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13633-018-0056-3","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/3/2 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
A novel, homozygous mutation in desert hedgehog (DHH) in a 46, XY patient with dysgenetic testes presenting with primary amenorrhoea: a case report.
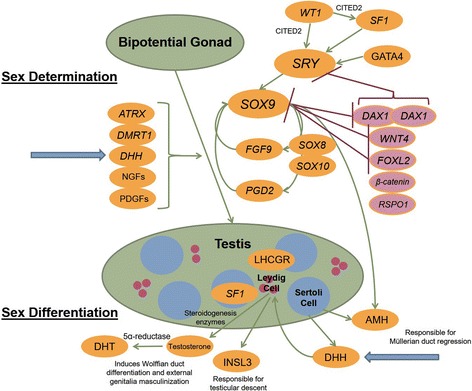
Background: Desert hedgehog (DHH) mutations have been described in only a limited number of individuals with 46, XY disorders of sex development (DSD) presenting as either partial or complete gonadal dysgenesis. Gonadal tumours and peripheral neuropathy have been associated with DHH mutations. Herein we report a novel, homozygous mutation of DHH identified through a targeted, massively parallel sequencing (MPS) DSD panel, in a patient presenting with partial gonadal dysgenesis. This novel mutation is two amino acids away from a previously described mutation in a patient who presented with complete gonadal dysgenesis. Adding to the complexity of work-up, our patient also expressed gender identity concern.
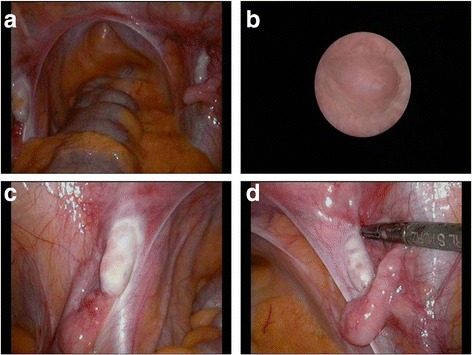
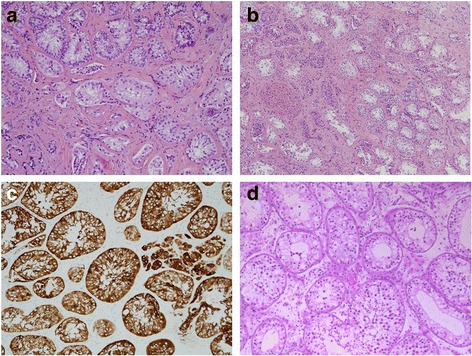
Case presentation: A 14-year-old, phenotypic female presented with primary amenorrhoea and absent secondary sex characteristics. Investigations revealed elevated gonadotrophins with low oestradiol, testosterone of 0.6 nmol/L and a 46, XY karyotype. Müllerian structures were not seen on pelvic ultrasound or laparoscopically and gonadal biopsies demonstrated dysgenetic testes without neoplasia (partial gonadal dysgenesis). The patient expressed gender identity confusion upon initial notification of investigation findings. Formal psychiatric evaluation excluded gender dysphoria. Genetic analysis was performed using a targeted, MPS DSD panel of 64 diagnostic and 927 research candidate genes. This identified a novel, homozygous mutation in exon 2 of DHH (DHH:NM_021044:exon2:c.G491C:p.R164P). With this finding our patient was screened for the possibility of peripheral neuropathy which was not evident clinically nor on investigation. She was commenced on oestrogen for pubertal induction.
Conclusion: The evaluation of patients with DSD is associated with considerable psychological distress. Targeted MPS enables an affordable and efficient method for diagnosis of 46, XY DSD cases. Identifying a genetic diagnosis may inform clinical management and in this case directed screening for peripheral neuropathy. In addition to the structural location of the mutation other interacting factors may influence phenotypic expression in homozygous DHH mutations.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


