Juan Sotos, Katherine Miller, Donald Corsmeier, Naomi Tokar, Benjamin Kelly, Vijay Nadella, Huachun Zhong, Amy Wetzel, Brent Adler, Chack-Yung Yu, Peter White
{"title":"van Maldergem综合征伴内分泌异常、促性腺功能减退、乳腺发育不全/发育不全1例。","authors":"Juan Sotos, Katherine Miller, Donald Corsmeier, Naomi Tokar, Benjamin Kelly, Vijay Nadella, Huachun Zhong, Amy Wetzel, Brent Adler, Chack-Yung Yu, Peter White","doi":"10.1186/s13633-017-0052-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>We report a female patient with endocrine abnormalities, hypogonadotropic hypogonadism and amazia (breasts aplasia/hypoplasia but normal nipples and areolas) in a rare syndrome: Van Maldergem syndrome (VMS).</p><p><strong>Case presentation: </strong>Our patient was first evaluated at age 4 for intellectual disability, craniofacial features, and auditory malformations. At age 15, she presented with no breast development and other findings consistent with hypogonadotropic hypogonadism. At age 37, she underwent whole exome sequencing (WES) to identify pathogenic variants. WES revealed compound heterozygous variants in <i>DCHS1</i> (rs145099391:G > A, p.P197L & rs753548138:G > A, <i>p</i>.T2334 M) [RefSeq NM_003737.3], diagnostic of Van Maldergem syndrome (VMS-1). VMS is a rare autosomal disorder reported in only 13 patients, characterized by intellectual disability, typical craniofacial features, auditory malformations, hearing loss, skeletal and limb malformations, brain abnormalities with periventricular neuronal heterotopia and other variable anomalies. Our patient had similar phenotypic abnormalities. She also had hypogonadotropic hypogonadism and amazia. Based on the clinical findings reported, two previously published patients with VMS may also have been affected by hypogonadotropic hypogonadism, but endocrine abnormalities were not evaluated or mentioned.</p><p><strong>Conclusion: </strong>This case highlights an individual with VMS, characterized by compound heterozygous variants in <i>DCHS1</i>. Our observations may provide additional information on the phenotypic spectrum of VMS, including hypogonadotropic hypogonadism and amazia. However, the molecular genetic basis for endocrine anomalies observed in some VMS patients, including ours, remains unexplained.</p>","PeriodicalId":14271,"journal":{"name":"International Journal of Pediatric Endocrinology","volume":"2017 ","pages":"12"},"PeriodicalIF":0.0000,"publicationDate":"2017-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13633-017-0052-z","citationCount":"5","resultStr":"{\"title\":\"A patient with van Maldergem syndrome with endocrine abnormalities, hypogonadotropic hypogonadism, and breast aplasia/hypoplasia.\",\"authors\":\"Juan Sotos, Katherine Miller, Donald Corsmeier, Naomi Tokar, Benjamin Kelly, Vijay Nadella, Huachun Zhong, Amy Wetzel, Brent Adler, Chack-Yung Yu, Peter White\",\"doi\":\"10.1186/s13633-017-0052-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>We report a female patient with endocrine abnormalities, hypogonadotropic hypogonadism and amazia (breasts aplasia/hypoplasia but normal nipples and areolas) in a rare syndrome: Van Maldergem syndrome (VMS).</p><p><strong>Case presentation: </strong>Our patient was first evaluated at age 4 for intellectual disability, craniofacial features, and auditory malformations. At age 15, she presented with no breast development and other findings consistent with hypogonadotropic hypogonadism. At age 37, she underwent whole exome sequencing (WES) to identify pathogenic variants. WES revealed compound heterozygous variants in <i>DCHS1</i> (rs145099391:G > A, p.P197L & rs753548138:G > A, <i>p</i>.T2334 M) [RefSeq NM_003737.3], diagnostic of Van Maldergem syndrome (VMS-1). VMS is a rare autosomal disorder reported in only 13 patients, characterized by intellectual disability, typical craniofacial features, auditory malformations, hearing loss, skeletal and limb malformations, brain abnormalities with periventricular neuronal heterotopia and other variable anomalies. Our patient had similar phenotypic abnormalities. She also had hypogonadotropic hypogonadism and amazia. Based on the clinical findings reported, two previously published patients with VMS may also have been affected by hypogonadotropic hypogonadism, but endocrine abnormalities were not evaluated or mentioned.</p><p><strong>Conclusion: </strong>This case highlights an individual with VMS, characterized by compound heterozygous variants in <i>DCHS1</i>. Our observations may provide additional information on the phenotypic spectrum of VMS, including hypogonadotropic hypogonadism and amazia. However, the molecular genetic basis for endocrine anomalies observed in some VMS patients, including ours, remains unexplained.</p>\",\"PeriodicalId\":14271,\"journal\":{\"name\":\"International Journal of Pediatric Endocrinology\",\"volume\":\"2017 \",\"pages\":\"12\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2017-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13633-017-0052-z\",\"citationCount\":\"5\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Pediatric Endocrinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13633-017-0052-z\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2017/10/13 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13633-017-0052-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2017/10/13 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 5
摘要
背景:我们报告了一位女性内分泌异常,促性腺功能低下和amazia(乳房发育不全/发育不全但乳头和乳晕正常)的罕见综合征:Van Maldergem综合征(VMS)。病例介绍:我们的患者在4岁时首次被评估为智力残疾,颅面特征和听觉畸形。15岁时,她没有乳房发育,其他表现符合促性腺功能减退症。37岁时,她接受了全外显子组测序(WES)来鉴定致病变异。WES检测发现DCHS1 (rs145099391:G > A, p.P197L和rs753548138:G > A, p.T2334 M) [RefSeq NM_003737.3]复合杂合变异体,诊断Van Maldergem综合征(VMS-1)。VMS是一种罕见的常染色体疾病,仅报道了13例患者,其特征为智力残疾,典型颅面特征,听觉畸形,听力丧失,骨骼和肢体畸形,脑异常伴脑室周围神经元异位和其他可变异常。我们的病人也有类似的表型异常。她还患有促性腺激素减退症和amazia。根据所报道的临床结果,先前发表的两例VMS患者也可能受到促性腺功能低下的影响,但内分泌异常未被评估或提及。结论:该病例突出了一个VMS个体,其特征是DCHS1的复合杂合变异体。我们的观察结果可能为VMS的表型谱提供额外的信息,包括促性腺功能低下和amazia。然而,在包括我们在内的一些VMS患者中观察到的内分泌异常的分子遗传学基础仍未得到解释。
A patient with van Maldergem syndrome with endocrine abnormalities, hypogonadotropic hypogonadism, and breast aplasia/hypoplasia.
Background: We report a female patient with endocrine abnormalities, hypogonadotropic hypogonadism and amazia (breasts aplasia/hypoplasia but normal nipples and areolas) in a rare syndrome: Van Maldergem syndrome (VMS).
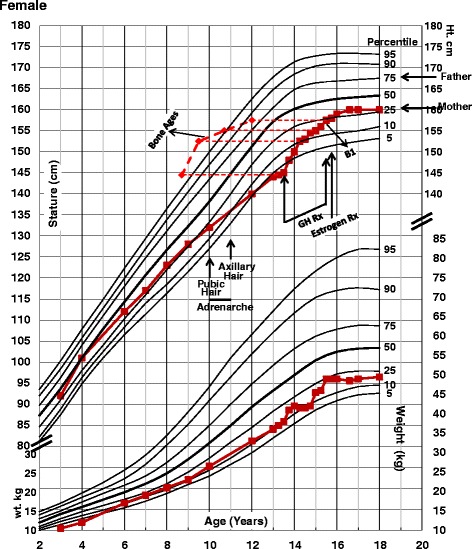
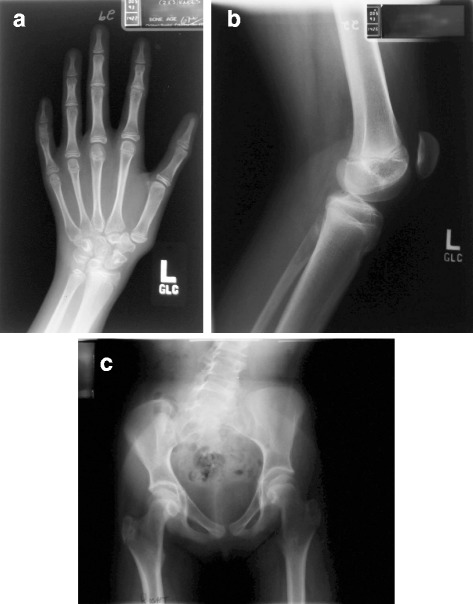
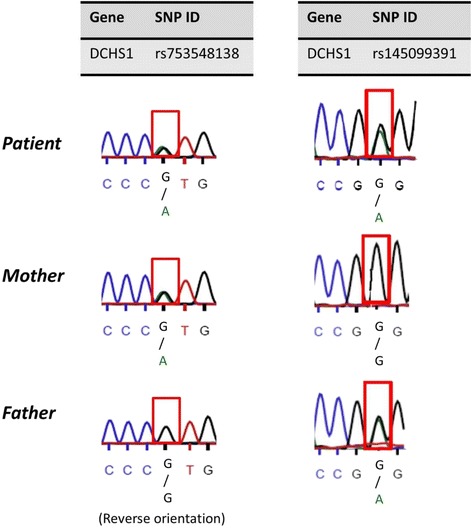
Case presentation: Our patient was first evaluated at age 4 for intellectual disability, craniofacial features, and auditory malformations. At age 15, she presented with no breast development and other findings consistent with hypogonadotropic hypogonadism. At age 37, she underwent whole exome sequencing (WES) to identify pathogenic variants. WES revealed compound heterozygous variants in DCHS1 (rs145099391:G > A, p.P197L & rs753548138:G > A, p.T2334 M) [RefSeq NM_003737.3], diagnostic of Van Maldergem syndrome (VMS-1). VMS is a rare autosomal disorder reported in only 13 patients, characterized by intellectual disability, typical craniofacial features, auditory malformations, hearing loss, skeletal and limb malformations, brain abnormalities with periventricular neuronal heterotopia and other variable anomalies. Our patient had similar phenotypic abnormalities. She also had hypogonadotropic hypogonadism and amazia. Based on the clinical findings reported, two previously published patients with VMS may also have been affected by hypogonadotropic hypogonadism, but endocrine abnormalities were not evaluated or mentioned.
Conclusion: This case highlights an individual with VMS, characterized by compound heterozygous variants in DCHS1. Our observations may provide additional information on the phenotypic spectrum of VMS, including hypogonadotropic hypogonadism and amazia. However, the molecular genetic basis for endocrine anomalies observed in some VMS patients, including ours, remains unexplained.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


