Y Sifi, M Medjroubi, R Froissart, N Taghane, K Sifi, A Benhabiles, S Lemai, S Semra, H Benmekhebi, Z Bouderda, N Abadi, A Hamri
{"title":"阿尔及利亚庞贝病临床分析。","authors":"Y Sifi, M Medjroubi, R Froissart, N Taghane, K Sifi, A Benhabiles, S Lemai, S Semra, H Benmekhebi, Z Bouderda, N Abadi, A Hamri","doi":"10.1155/2017/9427269","DOIUrl":null,"url":null,"abstract":"<p><p>Pompe's disease is a metabolic myopathy caused by a deficiency of acid alpha-glucosidase (GAA), also called acid maltase, an enzyme that degrades lysosomal glycogen. The clinical presentation of Pompe's disease is variable with respect to the age of onset and rate of disease progression. Patients with onset of symptoms in early infancy (infantile-onset Pompe disease (IOPD)) typically exhibit rapidly progressive hypertrophic cardiomyopathy and marked muscle weakness. Most of them die within the first year of life from cardiac and/or respiratory failure. In the majority of cases of Pompe's disease, onset of symptoms occurs after infancy, ranging widely from the first to sixth decade of life (late-onset Pompe's disease or LOPD). Progression of the disease is relentless and patients eventually progress to loss of ambulation and death due to respiratory failure. The objective of this study was to characterize the clinical presentation of 6 patients (3 with EOPD and the other 3 with LOPD) of 5 families from the East of Algeria. All our patients were diagnosed as having Pompe's disease based on biochemical confirmations of GAA deficiency by dried blood spots (DBS) and GAA gene mutations were analyzed in all patients who consented (<i>n</i> = 4). Our results are similar to other ethnic groups.</p>","PeriodicalId":16405,"journal":{"name":"Journal of Neurodegenerative Diseases","volume":"2017 ","pages":"9427269"},"PeriodicalIF":0.0000,"publicationDate":"2017-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2017/9427269","citationCount":"3","resultStr":"{\"title\":\"Clinical Analysis of Algerian Patients with Pompe Disease.\",\"authors\":\"Y Sifi, M Medjroubi, R Froissart, N Taghane, K Sifi, A Benhabiles, S Lemai, S Semra, H Benmekhebi, Z Bouderda, N Abadi, A Hamri\",\"doi\":\"10.1155/2017/9427269\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Pompe's disease is a metabolic myopathy caused by a deficiency of acid alpha-glucosidase (GAA), also called acid maltase, an enzyme that degrades lysosomal glycogen. The clinical presentation of Pompe's disease is variable with respect to the age of onset and rate of disease progression. Patients with onset of symptoms in early infancy (infantile-onset Pompe disease (IOPD)) typically exhibit rapidly progressive hypertrophic cardiomyopathy and marked muscle weakness. Most of them die within the first year of life from cardiac and/or respiratory failure. In the majority of cases of Pompe's disease, onset of symptoms occurs after infancy, ranging widely from the first to sixth decade of life (late-onset Pompe's disease or LOPD). Progression of the disease is relentless and patients eventually progress to loss of ambulation and death due to respiratory failure. The objective of this study was to characterize the clinical presentation of 6 patients (3 with EOPD and the other 3 with LOPD) of 5 families from the East of Algeria. All our patients were diagnosed as having Pompe's disease based on biochemical confirmations of GAA deficiency by dried blood spots (DBS) and GAA gene mutations were analyzed in all patients who consented (<i>n</i> = 4). Our results are similar to other ethnic groups.</p>\",\"PeriodicalId\":16405,\"journal\":{\"name\":\"Journal of Neurodegenerative Diseases\",\"volume\":\"2017 \",\"pages\":\"9427269\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2017-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1155/2017/9427269\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Neurodegenerative Diseases\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2017/9427269\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2017/2/6 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Neurodegenerative Diseases","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2017/9427269","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2017/2/6 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Clinical Analysis of Algerian Patients with Pompe Disease.
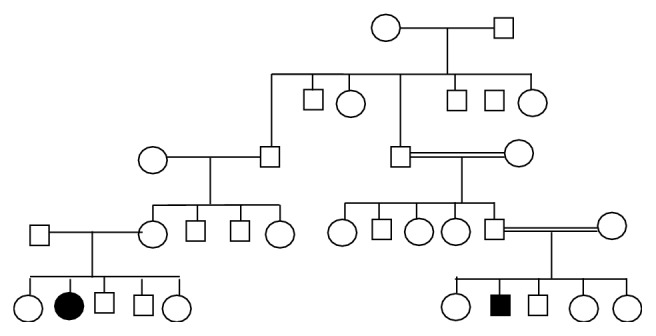
Pompe's disease is a metabolic myopathy caused by a deficiency of acid alpha-glucosidase (GAA), also called acid maltase, an enzyme that degrades lysosomal glycogen. The clinical presentation of Pompe's disease is variable with respect to the age of onset and rate of disease progression. Patients with onset of symptoms in early infancy (infantile-onset Pompe disease (IOPD)) typically exhibit rapidly progressive hypertrophic cardiomyopathy and marked muscle weakness. Most of them die within the first year of life from cardiac and/or respiratory failure. In the majority of cases of Pompe's disease, onset of symptoms occurs after infancy, ranging widely from the first to sixth decade of life (late-onset Pompe's disease or LOPD). Progression of the disease is relentless and patients eventually progress to loss of ambulation and death due to respiratory failure. The objective of this study was to characterize the clinical presentation of 6 patients (3 with EOPD and the other 3 with LOPD) of 5 families from the East of Algeria. All our patients were diagnosed as having Pompe's disease based on biochemical confirmations of GAA deficiency by dried blood spots (DBS) and GAA gene mutations were analyzed in all patients who consented (n = 4). Our results are similar to other ethnic groups.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


