Hiroto Tsujikawa, Kenta Sato, Cao Wei, Gul Saad, Kazuya Sumikoshi, Shugo Nakamura, Tohru Terada, Kentaro Shimizu
{"title":"基于相互作用能和序列守恒的蛋白质-配体结合位点预测方法的建立。","authors":"Hiroto Tsujikawa, Kenta Sato, Cao Wei, Gul Saad, Kazuya Sumikoshi, Shugo Nakamura, Tohru Terada, Kentaro Shimizu","doi":"10.1007/s10969-016-9204-2","DOIUrl":null,"url":null,"abstract":"<p><p>We present a new method for predicting protein-ligand-binding sites based on protein three-dimensional structure and amino acid conservation. This method involves calculation of the van der Waals interaction energy between a protein and many probes placed on the protein surface and subsequent clustering of the probes with low interaction energies to identify the most energetically favorable locus. In addition, it uses amino acid conservation among homologous proteins. Ligand-binding sites were predicted by combining the interaction energy and the amino acid conservation score. The performance of our prediction method was evaluated using a non-redundant dataset of 348 ligand-bound and ligand-unbound protein structure pairs, constructed by filtering entries in a ligand-binding site structure database, LigASite. Ligand-bound structure prediction (bound prediction) indicated that 74.0 % of predicted ligand-binding sites overlapped with real ligand-binding sites by over 25 % of their volume. Ligand-unbound structure prediction (unbound prediction) indicated that 73.9 % of predicted ligand-binding residues overlapped with real ligand-binding residues. The amino acid conservation score improved the average prediction accuracy by 17.0 and 17.6 points for the bound and unbound predictions, respectively. These results demonstrate the effectiveness of the combined use of the interaction energy and amino acid conservation in the ligand-binding site prediction. </p>","PeriodicalId":73957,"journal":{"name":"Journal of structural and functional genomics","volume":"17 2-3","pages":"39-49"},"PeriodicalIF":0.0000,"publicationDate":"2016-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1007/s10969-016-9204-2","citationCount":"10","resultStr":"{\"title\":\"Development of a protein-ligand-binding site prediction method based on interaction energy and sequence conservation.\",\"authors\":\"Hiroto Tsujikawa, Kenta Sato, Cao Wei, Gul Saad, Kazuya Sumikoshi, Shugo Nakamura, Tohru Terada, Kentaro Shimizu\",\"doi\":\"10.1007/s10969-016-9204-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>We present a new method for predicting protein-ligand-binding sites based on protein three-dimensional structure and amino acid conservation. This method involves calculation of the van der Waals interaction energy between a protein and many probes placed on the protein surface and subsequent clustering of the probes with low interaction energies to identify the most energetically favorable locus. In addition, it uses amino acid conservation among homologous proteins. Ligand-binding sites were predicted by combining the interaction energy and the amino acid conservation score. The performance of our prediction method was evaluated using a non-redundant dataset of 348 ligand-bound and ligand-unbound protein structure pairs, constructed by filtering entries in a ligand-binding site structure database, LigASite. Ligand-bound structure prediction (bound prediction) indicated that 74.0 % of predicted ligand-binding sites overlapped with real ligand-binding sites by over 25 % of their volume. Ligand-unbound structure prediction (unbound prediction) indicated that 73.9 % of predicted ligand-binding residues overlapped with real ligand-binding residues. The amino acid conservation score improved the average prediction accuracy by 17.0 and 17.6 points for the bound and unbound predictions, respectively. These results demonstrate the effectiveness of the combined use of the interaction energy and amino acid conservation in the ligand-binding site prediction. </p>\",\"PeriodicalId\":73957,\"journal\":{\"name\":\"Journal of structural and functional genomics\",\"volume\":\"17 2-3\",\"pages\":\"39-49\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2016-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1007/s10969-016-9204-2\",\"citationCount\":\"10\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of structural and functional genomics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1007/s10969-016-9204-2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/7/11 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of structural and functional genomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s10969-016-9204-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/7/11 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Development of a protein-ligand-binding site prediction method based on interaction energy and sequence conservation.
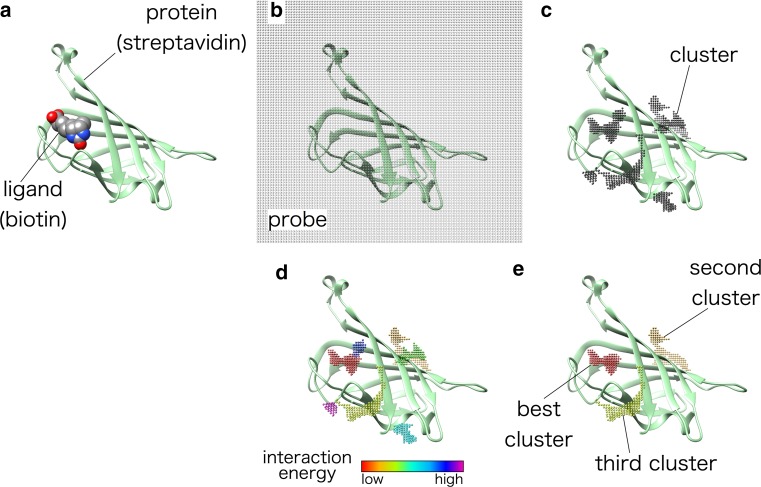
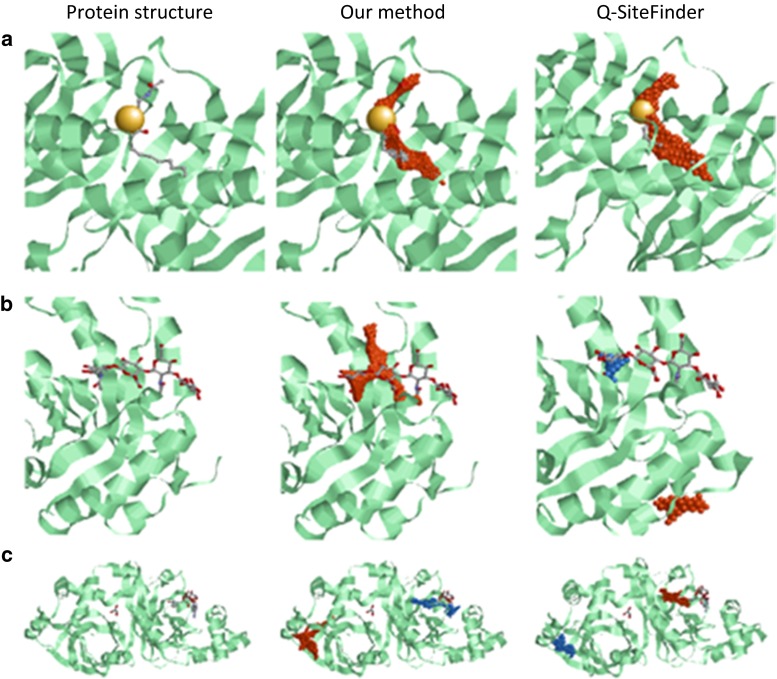
We present a new method for predicting protein-ligand-binding sites based on protein three-dimensional structure and amino acid conservation. This method involves calculation of the van der Waals interaction energy between a protein and many probes placed on the protein surface and subsequent clustering of the probes with low interaction energies to identify the most energetically favorable locus. In addition, it uses amino acid conservation among homologous proteins. Ligand-binding sites were predicted by combining the interaction energy and the amino acid conservation score. The performance of our prediction method was evaluated using a non-redundant dataset of 348 ligand-bound and ligand-unbound protein structure pairs, constructed by filtering entries in a ligand-binding site structure database, LigASite. Ligand-bound structure prediction (bound prediction) indicated that 74.0 % of predicted ligand-binding sites overlapped with real ligand-binding sites by over 25 % of their volume. Ligand-unbound structure prediction (unbound prediction) indicated that 73.9 % of predicted ligand-binding residues overlapped with real ligand-binding residues. The amino acid conservation score improved the average prediction accuracy by 17.0 and 17.6 points for the bound and unbound predictions, respectively. These results demonstrate the effectiveness of the combined use of the interaction energy and amino acid conservation in the ligand-binding site prediction.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


