Ioannis Valavanis, Eleftherios Pilalis, Panagiotis Georgiadis, Soterios Kyrtopoulos, Aristotelis Chatziioannou
{"title":"基因组尺度DNA甲基化的癌症生物标志物:进化和语义分析方法的比较。","authors":"Ioannis Valavanis, Eleftherios Pilalis, Panagiotis Georgiadis, Soterios Kyrtopoulos, Aristotelis Chatziioannou","doi":"10.3390/microarrays4040647","DOIUrl":null,"url":null,"abstract":"<p><p>DNA methylation profiling exploits microarray technologies, thus yielding a wealth of high-volume data. Here, an intelligent framework is applied, encompassing epidemiological genome-scale DNA methylation data produced from the Illumina's Infinium Human Methylation 450K Bead Chip platform, in an effort to correlate interesting methylation patterns with cancer predisposition and, in particular, breast cancer and B-cell lymphoma. Feature selection and classification are employed in order to select, from an initial set of ~480,000 methylation measurements at CpG sites, predictive cancer epigenetic biomarkers and assess their classification power for discriminating healthy versus cancer related classes. Feature selection exploits evolutionary algorithms or a graph-theoretic methodology which makes use of the semantics information included in the Gene Ontology (GO) tree. The selected features, corresponding to methylation of CpG sites, attained moderate-to-high classification accuracies when imported to a series of classifiers evaluated by resampling or blindfold validation. The semantics-driven selection revealed sets of CpG sites performing similarly with evolutionary selection in the classification tasks. However, gene enrichment and pathway analysis showed that it additionally provides more descriptive sets of GO terms and KEGG pathways regarding the cancer phenotypes studied here. Results support the expediency of this methodology regarding its application in epidemiological studies. </p>","PeriodicalId":56355,"journal":{"name":"Microarrays","volume":"4 4","pages":"647-70"},"PeriodicalIF":0.0000,"publicationDate":"2015-11-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.3390/microarrays4040647","citationCount":"7","resultStr":"{\"title\":\"Cancer Biomarkers from Genome-Scale DNA Methylation: Comparison of Evolutionary and Semantic Analysis Methods.\",\"authors\":\"Ioannis Valavanis, Eleftherios Pilalis, Panagiotis Georgiadis, Soterios Kyrtopoulos, Aristotelis Chatziioannou\",\"doi\":\"10.3390/microarrays4040647\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>DNA methylation profiling exploits microarray technologies, thus yielding a wealth of high-volume data. Here, an intelligent framework is applied, encompassing epidemiological genome-scale DNA methylation data produced from the Illumina's Infinium Human Methylation 450K Bead Chip platform, in an effort to correlate interesting methylation patterns with cancer predisposition and, in particular, breast cancer and B-cell lymphoma. Feature selection and classification are employed in order to select, from an initial set of ~480,000 methylation measurements at CpG sites, predictive cancer epigenetic biomarkers and assess their classification power for discriminating healthy versus cancer related classes. Feature selection exploits evolutionary algorithms or a graph-theoretic methodology which makes use of the semantics information included in the Gene Ontology (GO) tree. The selected features, corresponding to methylation of CpG sites, attained moderate-to-high classification accuracies when imported to a series of classifiers evaluated by resampling or blindfold validation. The semantics-driven selection revealed sets of CpG sites performing similarly with evolutionary selection in the classification tasks. However, gene enrichment and pathway analysis showed that it additionally provides more descriptive sets of GO terms and KEGG pathways regarding the cancer phenotypes studied here. Results support the expediency of this methodology regarding its application in epidemiological studies. </p>\",\"PeriodicalId\":56355,\"journal\":{\"name\":\"Microarrays\",\"volume\":\"4 4\",\"pages\":\"647-70\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2015-11-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.3390/microarrays4040647\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Microarrays\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/microarrays4040647\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microarrays","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/microarrays4040647","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Cancer Biomarkers from Genome-Scale DNA Methylation: Comparison of Evolutionary and Semantic Analysis Methods.
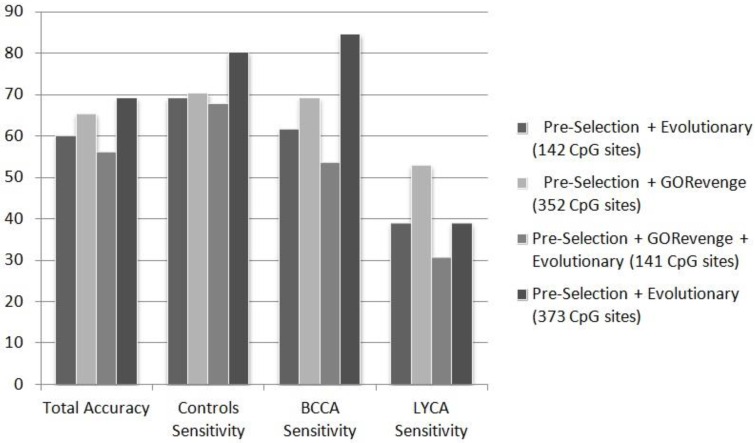
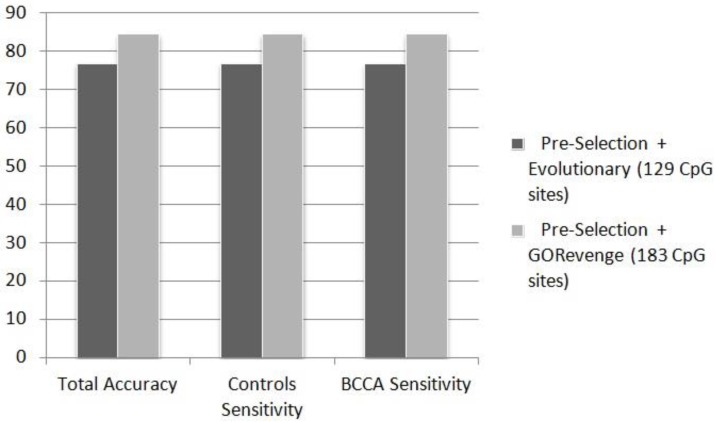
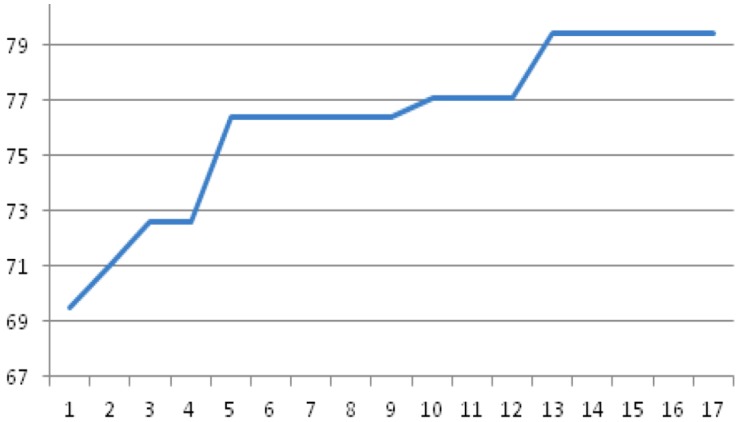
DNA methylation profiling exploits microarray technologies, thus yielding a wealth of high-volume data. Here, an intelligent framework is applied, encompassing epidemiological genome-scale DNA methylation data produced from the Illumina's Infinium Human Methylation 450K Bead Chip platform, in an effort to correlate interesting methylation patterns with cancer predisposition and, in particular, breast cancer and B-cell lymphoma. Feature selection and classification are employed in order to select, from an initial set of ~480,000 methylation measurements at CpG sites, predictive cancer epigenetic biomarkers and assess their classification power for discriminating healthy versus cancer related classes. Feature selection exploits evolutionary algorithms or a graph-theoretic methodology which makes use of the semantics information included in the Gene Ontology (GO) tree. The selected features, corresponding to methylation of CpG sites, attained moderate-to-high classification accuracies when imported to a series of classifiers evaluated by resampling or blindfold validation. The semantics-driven selection revealed sets of CpG sites performing similarly with evolutionary selection in the classification tasks. However, gene enrichment and pathway analysis showed that it additionally provides more descriptive sets of GO terms and KEGG pathways regarding the cancer phenotypes studied here. Results support the expediency of this methodology regarding its application in epidemiological studies.
期刊介绍:
High-Throughput (formerly Microarrays, ISSN 2076-3905) is a multidisciplinary peer-reviewed scientific journal that provides an advanced forum for the publication of studies reporting high-dimensional approaches and developments in Life Sciences, Chemistry and related fields. Our aim is to encourage scientists to publish their experimental and theoretical results based on high-throughput techniques as well as computational and statistical tools for data analysis and interpretation. The full experimental or methodological details must be provided so that the results can be reproduced. There is no restriction on the length of the papers. High-Throughput invites submissions covering several topics, including, but not limited to: Microarrays, DNA Sequencing, RNA Sequencing, Protein Identification and Quantification, Cell-based Approaches, Omics Technologies, Imaging, Bioinformatics, Computational Biology/Chemistry, Statistics, Integrative Omics, Drug Discovery and Development, Microfluidics, Lab-on-a-chip, Data Mining, Databases, Multiplex Assays.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


