Ana Barat, Heather J Ruskin, Annette T Byrne, Jochen H M Prehn
{"title":"整合结肠癌微阵列数据:将基因座特异性甲基化组与基于基因表达的分类相关联。","authors":"Ana Barat, Heather J Ruskin, Annette T Byrne, Jochen H M Prehn","doi":"10.3390/microarrays4040630","DOIUrl":null,"url":null,"abstract":"<p><p>Recently, considerable attention has been paid to gene expression-based classifications of colorectal cancers (CRC) and their association with patient prognosis. In addition to changes in gene expression, abnormal DNA-methylation is known to play an important role in cancer onset and development, and colon cancer is no exception to this rule. Large-scale technologies, such as methylation microarray assays and specific sequencing of methylated DNA, have been used to determine whole genome profiles of CpG island methylation in tissue samples. In this article, publicly available microarray-based gene expression and methylation data sets are used to characterize expression subtypes with respect to locus-specific methylation. A major objective was to determine whether integration of these data types improves previously characterized subtypes, or provides evidence for additional subtypes. We used unsupervised clustering techniques to determine methylation-based subgroups, which are subsequently annotated with three published expression-based classifications, comprising from three to six subtypes. Our results showed that, while methylation profiles provide a further basis for segregation of certain (Inflammatory and Goblet-like) finer-grained expression-based subtypes, they also suggest that other finer-grained subtypes are not distinctive and can be considered as a single subtype. </p>","PeriodicalId":56355,"journal":{"name":"Microarrays","volume":"4 4","pages":"630-46"},"PeriodicalIF":0.0000,"publicationDate":"2015-11-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.3390/microarrays4040630","citationCount":"3","resultStr":"{\"title\":\"Integrating Colon Cancer Microarray Data: Associating Locus-Specific Methylation Groups to Gene Expression-Based Classifications.\",\"authors\":\"Ana Barat, Heather J Ruskin, Annette T Byrne, Jochen H M Prehn\",\"doi\":\"10.3390/microarrays4040630\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Recently, considerable attention has been paid to gene expression-based classifications of colorectal cancers (CRC) and their association with patient prognosis. In addition to changes in gene expression, abnormal DNA-methylation is known to play an important role in cancer onset and development, and colon cancer is no exception to this rule. Large-scale technologies, such as methylation microarray assays and specific sequencing of methylated DNA, have been used to determine whole genome profiles of CpG island methylation in tissue samples. In this article, publicly available microarray-based gene expression and methylation data sets are used to characterize expression subtypes with respect to locus-specific methylation. A major objective was to determine whether integration of these data types improves previously characterized subtypes, or provides evidence for additional subtypes. We used unsupervised clustering techniques to determine methylation-based subgroups, which are subsequently annotated with three published expression-based classifications, comprising from three to six subtypes. Our results showed that, while methylation profiles provide a further basis for segregation of certain (Inflammatory and Goblet-like) finer-grained expression-based subtypes, they also suggest that other finer-grained subtypes are not distinctive and can be considered as a single subtype. </p>\",\"PeriodicalId\":56355,\"journal\":{\"name\":\"Microarrays\",\"volume\":\"4 4\",\"pages\":\"630-46\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2015-11-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.3390/microarrays4040630\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Microarrays\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/microarrays4040630\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microarrays","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/microarrays4040630","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Integrating Colon Cancer Microarray Data: Associating Locus-Specific Methylation Groups to Gene Expression-Based Classifications.
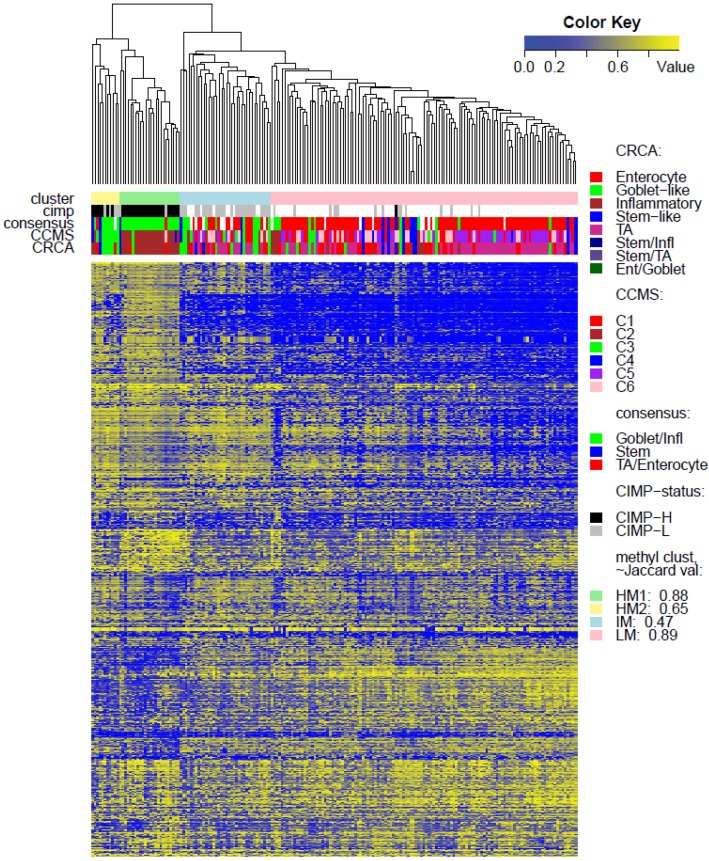
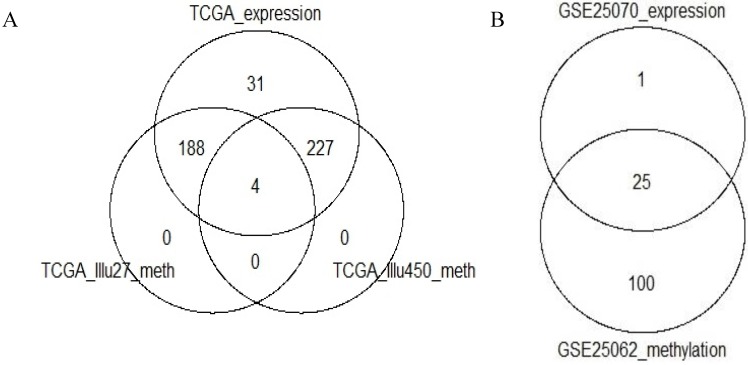
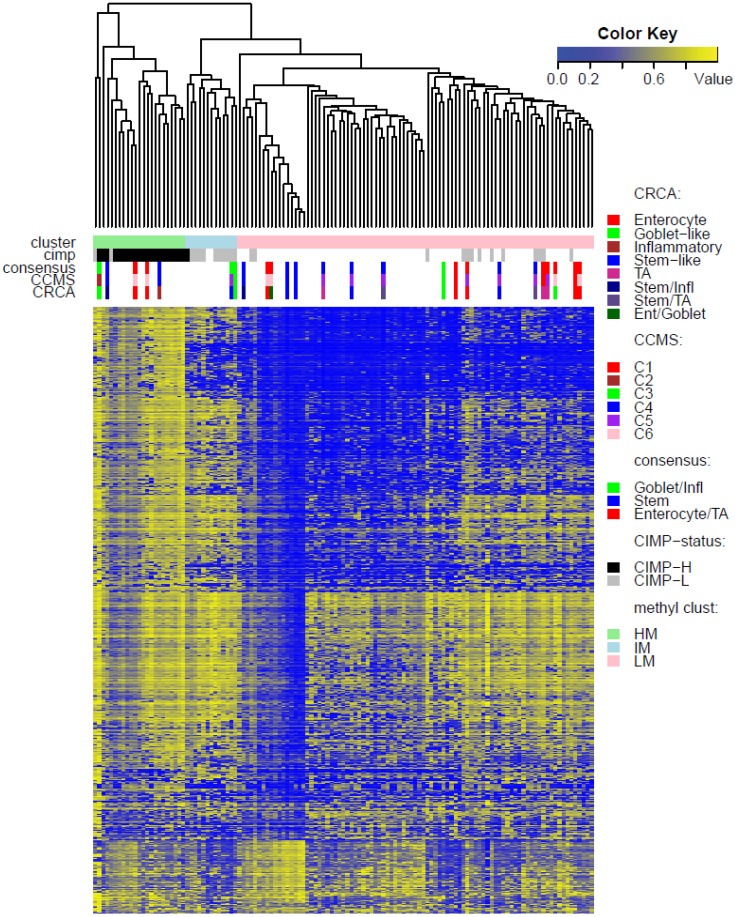
Recently, considerable attention has been paid to gene expression-based classifications of colorectal cancers (CRC) and their association with patient prognosis. In addition to changes in gene expression, abnormal DNA-methylation is known to play an important role in cancer onset and development, and colon cancer is no exception to this rule. Large-scale technologies, such as methylation microarray assays and specific sequencing of methylated DNA, have been used to determine whole genome profiles of CpG island methylation in tissue samples. In this article, publicly available microarray-based gene expression and methylation data sets are used to characterize expression subtypes with respect to locus-specific methylation. A major objective was to determine whether integration of these data types improves previously characterized subtypes, or provides evidence for additional subtypes. We used unsupervised clustering techniques to determine methylation-based subgroups, which are subsequently annotated with three published expression-based classifications, comprising from three to six subtypes. Our results showed that, while methylation profiles provide a further basis for segregation of certain (Inflammatory and Goblet-like) finer-grained expression-based subtypes, they also suggest that other finer-grained subtypes are not distinctive and can be considered as a single subtype.
期刊介绍:
High-Throughput (formerly Microarrays, ISSN 2076-3905) is a multidisciplinary peer-reviewed scientific journal that provides an advanced forum for the publication of studies reporting high-dimensional approaches and developments in Life Sciences, Chemistry and related fields. Our aim is to encourage scientists to publish their experimental and theoretical results based on high-throughput techniques as well as computational and statistical tools for data analysis and interpretation. The full experimental or methodological details must be provided so that the results can be reproduced. There is no restriction on the length of the papers. High-Throughput invites submissions covering several topics, including, but not limited to: Microarrays, DNA Sequencing, RNA Sequencing, Protein Identification and Quantification, Cell-based Approaches, Omics Technologies, Imaging, Bioinformatics, Computational Biology/Chemistry, Statistics, Integrative Omics, Drug Discovery and Development, Microfluidics, Lab-on-a-chip, Data Mining, Databases, Multiplex Assays.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


