{"title":"具有Klinefelter综合征临床特征的罕见47xxy / 46xx嵌合。","authors":"Noor Shafina Mohd Nor, Muhammad Yazid Jalaludin","doi":"10.1186/s13633-016-0029-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>47 XXY/46 XX mosaicism with characteristics suggesting Klinefelter syndrome is very rare and at present, only seven cases have been reported in the literature.</p><p><strong>Case presentation: </strong>We report an Indian boy diagnosed as variant of Klinefelter syndrome with 47 XXY/46 XX mosaicism at age 12 years. He was noted to have right cryptorchidism and chordae at birth, but did not have surgery for these until age 3 years. During surgery, the right gonad was atrophic and removed. Histology revealed atrophic ovarian tissue. Pelvic ultrasound showed no Mullerian structures. There was however no clinical follow up and he was raised as a boy. At 12 years old he was re-evaluated because of parental concern about his 'female' body habitus. He was slightly overweight, had eunuchoid body habitus with mild gynaecomastia. The right scrotal sac was empty and a 2mls testis was present in the left scrotum. Penile length was 5.2 cm and width 2.0 cm. There was absent pubic or axillary hair. Pronation and supination of his upper limbs were reduced and x-ray of both elbow joints revealed bilateral radioulnar synostosis. The baseline laboratory data were LH < 0.1 mIU/ml, FSH 1.4 mIU/ml, testosterone 0.6 nmol/L with raised estradiol, 96 pmol/L. HCG stimulation test showed poor Leydig cell response. The karyotype based on 76 cells was 47 XXY[9]/46 XX[67] with SRY positive. Laparoscopic examination revealed no Mullerian structures.</p><p><strong>Conclusion: </strong>Insisting on an adequate number of cells (at least 50) to be examined during karyotyping is important so as not to miss diagnosing mosaicism.</p>","PeriodicalId":14271,"journal":{"name":"International Journal of Pediatric Endocrinology","volume":null,"pages":null},"PeriodicalIF":0.0000,"publicationDate":"2016-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13633-016-0029-3","citationCount":"18","resultStr":"{\"title\":\"A rare 47 XXY/46 XX mosaicism with clinical features of Klinefelter syndrome.\",\"authors\":\"Noor Shafina Mohd Nor, Muhammad Yazid Jalaludin\",\"doi\":\"10.1186/s13633-016-0029-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>47 XXY/46 XX mosaicism with characteristics suggesting Klinefelter syndrome is very rare and at present, only seven cases have been reported in the literature.</p><p><strong>Case presentation: </strong>We report an Indian boy diagnosed as variant of Klinefelter syndrome with 47 XXY/46 XX mosaicism at age 12 years. He was noted to have right cryptorchidism and chordae at birth, but did not have surgery for these until age 3 years. During surgery, the right gonad was atrophic and removed. Histology revealed atrophic ovarian tissue. Pelvic ultrasound showed no Mullerian structures. There was however no clinical follow up and he was raised as a boy. At 12 years old he was re-evaluated because of parental concern about his 'female' body habitus. He was slightly overweight, had eunuchoid body habitus with mild gynaecomastia. The right scrotal sac was empty and a 2mls testis was present in the left scrotum. Penile length was 5.2 cm and width 2.0 cm. There was absent pubic or axillary hair. Pronation and supination of his upper limbs were reduced and x-ray of both elbow joints revealed bilateral radioulnar synostosis. The baseline laboratory data were LH < 0.1 mIU/ml, FSH 1.4 mIU/ml, testosterone 0.6 nmol/L with raised estradiol, 96 pmol/L. HCG stimulation test showed poor Leydig cell response. The karyotype based on 76 cells was 47 XXY[9]/46 XX[67] with SRY positive. Laparoscopic examination revealed no Mullerian structures.</p><p><strong>Conclusion: </strong>Insisting on an adequate number of cells (at least 50) to be examined during karyotyping is important so as not to miss diagnosing mosaicism.</p>\",\"PeriodicalId\":14271,\"journal\":{\"name\":\"International Journal of Pediatric Endocrinology\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2016-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13633-016-0029-3\",\"citationCount\":\"18\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Pediatric Endocrinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13633-016-0029-3\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/6/2 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13633-016-0029-3","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/6/2 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
A rare 47 XXY/46 XX mosaicism with clinical features of Klinefelter syndrome.
Background: 47 XXY/46 XX mosaicism with characteristics suggesting Klinefelter syndrome is very rare and at present, only seven cases have been reported in the literature.
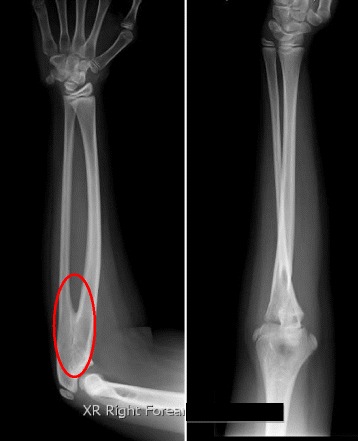
Case presentation: We report an Indian boy diagnosed as variant of Klinefelter syndrome with 47 XXY/46 XX mosaicism at age 12 years. He was noted to have right cryptorchidism and chordae at birth, but did not have surgery for these until age 3 years. During surgery, the right gonad was atrophic and removed. Histology revealed atrophic ovarian tissue. Pelvic ultrasound showed no Mullerian structures. There was however no clinical follow up and he was raised as a boy. At 12 years old he was re-evaluated because of parental concern about his 'female' body habitus. He was slightly overweight, had eunuchoid body habitus with mild gynaecomastia. The right scrotal sac was empty and a 2mls testis was present in the left scrotum. Penile length was 5.2 cm and width 2.0 cm. There was absent pubic or axillary hair. Pronation and supination of his upper limbs were reduced and x-ray of both elbow joints revealed bilateral radioulnar synostosis. The baseline laboratory data were LH < 0.1 mIU/ml, FSH 1.4 mIU/ml, testosterone 0.6 nmol/L with raised estradiol, 96 pmol/L. HCG stimulation test showed poor Leydig cell response. The karyotype based on 76 cells was 47 XXY[9]/46 XX[67] with SRY positive. Laparoscopic examination revealed no Mullerian structures.
Conclusion: Insisting on an adequate number of cells (at least 50) to be examined during karyotyping is important so as not to miss diagnosing mosaicism.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


