Luis A Cea, Jorge A Bevilacqua, Christian Arriagada, Ana María Cárdenas, Anne Bigot, Vincent Mouly, Juan C Sáez, Pablo Caviedes
{"title":"dysferlin缺失诱导人肌管中功能性连接蛋白半通道的表达。","authors":"Luis A Cea, Jorge A Bevilacqua, Christian Arriagada, Ana María Cárdenas, Anne Bigot, Vincent Mouly, Juan C Sáez, Pablo Caviedes","doi":"10.1186/s12860-016-0096-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Mutations in the gene encoding for dysferlin cause recessive autosomal muscular dystrophies called dysferlinopathies. These mutations induce several alterations in skeletal muscles, including, inflammation, increased membrane permeability and cell death. Despite the fact that the etiology of dysferlinopathies is known, the mechanism that explains the aforementioned alterations is still elusive. Therefore, we have now evaluated the potential involvement of connexin based hemichannels in the pathophysiology of dysferlinopathies.</p><p><strong>Results: </strong>Human deltoid muscle biopsies of 5 Chilean dysferlinopathy patients exhibited the presence of muscular connexins (Cx40.1, Cx43 and Cx45). The presence of these connexins was also observed in human myotubes derived from immortalized myoblasts derived from other patients with mutated forms of dysferlin. In addition to the aforementioned connexins, these myotubes expressed functional connexin based hemichannels, evaluated by ethidium uptake assays, as opposed to myotubes obtained from a normal human muscle cell line, RCMH. This response was reproduced in a knock-down model of dysferlin, by treating RCMH cell line with small hairpin RNA specific for dysferlin (RCMH-sh Dysferlin). Also, the presence of P2X7 receptor and the transient receptor potential channel, TRPV2, another Ca(2+) permeable channels, was detected in the myotubes expressing mutated dysferlin, and an elevated resting intracellular Ca(2+) level was found in the latter myotubes, which was in turn reduced to control levels in the presence of the molecule D4, a selective Cx HCs inhibitor.</p><p><strong>Conclusions: </strong>The data suggests that dysferlin deficiency, caused by mutation or downregulation of dysferlin, promotes the expression of Cx HCs. Then, the de novo expression Cx HC causes a dysregulation of intracellular free Ca(2+) levels, which could underlie muscular damage associated to dysferlin mutations. This mechanism could constitute a potential therapeutical target in dysferlinopathies.</p>","PeriodicalId":9051,"journal":{"name":"BMC Cell Biology","volume":"17 Suppl 1 ","pages":"15"},"PeriodicalIF":0.0000,"publicationDate":"2016-05-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12860-016-0096-6","citationCount":"15","resultStr":"{\"title\":\"The absence of dysferlin induces the expression of functional connexin-based hemichannels in human myotubes.\",\"authors\":\"Luis A Cea, Jorge A Bevilacqua, Christian Arriagada, Ana María Cárdenas, Anne Bigot, Vincent Mouly, Juan C Sáez, Pablo Caviedes\",\"doi\":\"10.1186/s12860-016-0096-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Mutations in the gene encoding for dysferlin cause recessive autosomal muscular dystrophies called dysferlinopathies. These mutations induce several alterations in skeletal muscles, including, inflammation, increased membrane permeability and cell death. Despite the fact that the etiology of dysferlinopathies is known, the mechanism that explains the aforementioned alterations is still elusive. Therefore, we have now evaluated the potential involvement of connexin based hemichannels in the pathophysiology of dysferlinopathies.</p><p><strong>Results: </strong>Human deltoid muscle biopsies of 5 Chilean dysferlinopathy patients exhibited the presence of muscular connexins (Cx40.1, Cx43 and Cx45). The presence of these connexins was also observed in human myotubes derived from immortalized myoblasts derived from other patients with mutated forms of dysferlin. In addition to the aforementioned connexins, these myotubes expressed functional connexin based hemichannels, evaluated by ethidium uptake assays, as opposed to myotubes obtained from a normal human muscle cell line, RCMH. This response was reproduced in a knock-down model of dysferlin, by treating RCMH cell line with small hairpin RNA specific for dysferlin (RCMH-sh Dysferlin). Also, the presence of P2X7 receptor and the transient receptor potential channel, TRPV2, another Ca(2+) permeable channels, was detected in the myotubes expressing mutated dysferlin, and an elevated resting intracellular Ca(2+) level was found in the latter myotubes, which was in turn reduced to control levels in the presence of the molecule D4, a selective Cx HCs inhibitor.</p><p><strong>Conclusions: </strong>The data suggests that dysferlin deficiency, caused by mutation or downregulation of dysferlin, promotes the expression of Cx HCs. Then, the de novo expression Cx HC causes a dysregulation of intracellular free Ca(2+) levels, which could underlie muscular damage associated to dysferlin mutations. This mechanism could constitute a potential therapeutical target in dysferlinopathies.</p>\",\"PeriodicalId\":9051,\"journal\":{\"name\":\"BMC Cell Biology\",\"volume\":\"17 Suppl 1 \",\"pages\":\"15\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2016-05-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12860-016-0096-6\",\"citationCount\":\"15\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Cell Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s12860-016-0096-6\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Cell Biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12860-016-0096-6","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
The absence of dysferlin induces the expression of functional connexin-based hemichannels in human myotubes.
Background: Mutations in the gene encoding for dysferlin cause recessive autosomal muscular dystrophies called dysferlinopathies. These mutations induce several alterations in skeletal muscles, including, inflammation, increased membrane permeability and cell death. Despite the fact that the etiology of dysferlinopathies is known, the mechanism that explains the aforementioned alterations is still elusive. Therefore, we have now evaluated the potential involvement of connexin based hemichannels in the pathophysiology of dysferlinopathies.
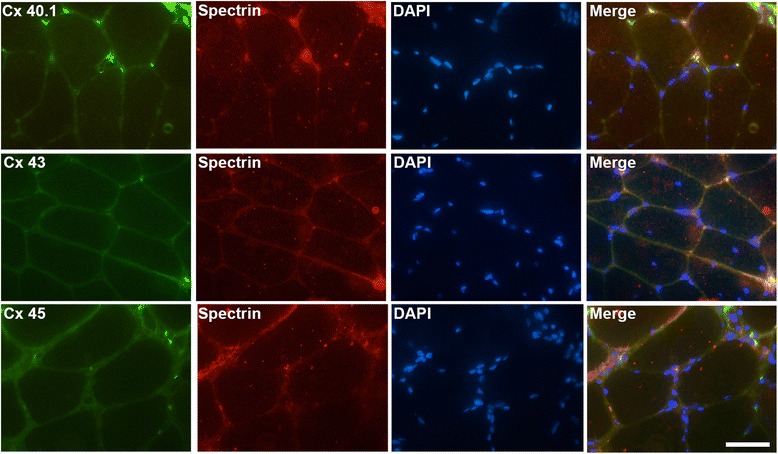
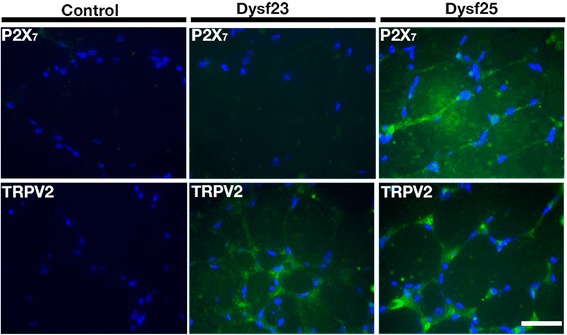
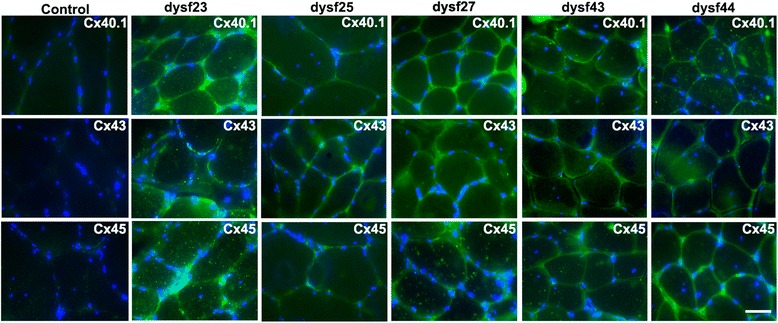
Results: Human deltoid muscle biopsies of 5 Chilean dysferlinopathy patients exhibited the presence of muscular connexins (Cx40.1, Cx43 and Cx45). The presence of these connexins was also observed in human myotubes derived from immortalized myoblasts derived from other patients with mutated forms of dysferlin. In addition to the aforementioned connexins, these myotubes expressed functional connexin based hemichannels, evaluated by ethidium uptake assays, as opposed to myotubes obtained from a normal human muscle cell line, RCMH. This response was reproduced in a knock-down model of dysferlin, by treating RCMH cell line with small hairpin RNA specific for dysferlin (RCMH-sh Dysferlin). Also, the presence of P2X7 receptor and the transient receptor potential channel, TRPV2, another Ca(2+) permeable channels, was detected in the myotubes expressing mutated dysferlin, and an elevated resting intracellular Ca(2+) level was found in the latter myotubes, which was in turn reduced to control levels in the presence of the molecule D4, a selective Cx HCs inhibitor.
Conclusions: The data suggests that dysferlin deficiency, caused by mutation or downregulation of dysferlin, promotes the expression of Cx HCs. Then, the de novo expression Cx HC causes a dysregulation of intracellular free Ca(2+) levels, which could underlie muscular damage associated to dysferlin mutations. This mechanism could constitute a potential therapeutical target in dysferlinopathies.
期刊介绍:
BMC Molecular and Cell Biology, formerly known as BMC Cell Biology, is an open access journal that considers articles on all aspects of both eukaryotic and prokaryotic cell and molecular biology, including structural and functional cell biology, DNA and RNA in a cellular context and biochemistry, as well as research using both the experimental and theoretical aspects of physics to study biological processes and investigations into the structure of biological macromolecules.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


