{"title":"途径分析在鉴别非小细胞肺癌潜在药物靶点中的稳健性。","authors":"Andrew Dalby, Ian Bailey","doi":"10.3390/microarrays3040212","DOIUrl":null,"url":null,"abstract":"<p><p>The identification of genes responsible for causing cancers from gene expression data has had varied success. Often the genes identified depend on the methods used for detecting expression patterns, or on the ways that the data had been normalized and filtered. The use of gene set enrichment analysis is one way to introduce biological information in order to improve the detection of differentially expressed genes and pathways. In this paper we show that the use of network models while still subject to the problems of normalization is a more robust method for detecting pathways that are differentially overrepresented in lung cancer data. Such differences may provide opportunities for novel therapeutics. In addition, we present evidence that non-small cell lung carcinoma is not a series of homogeneous diseases; rather that there is a heterogeny within the genotype which defies phenotype classification. This diversity helps to explain the lack of progress in developing therapies against non-small cell carcinoma and suggests that drug development may consider multiple pathways as treatment targets. </p>","PeriodicalId":56355,"journal":{"name":"Microarrays","volume":"3 4","pages":"212-25"},"PeriodicalIF":0.0000,"publicationDate":"2014-10-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.3390/microarrays3040212","citationCount":"1","resultStr":"{\"title\":\"The Robustness of Pathway Analysis in Identifying Potential Drug Targets in Non-Small Cell Lung Carcinoma.\",\"authors\":\"Andrew Dalby, Ian Bailey\",\"doi\":\"10.3390/microarrays3040212\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The identification of genes responsible for causing cancers from gene expression data has had varied success. Often the genes identified depend on the methods used for detecting expression patterns, or on the ways that the data had been normalized and filtered. The use of gene set enrichment analysis is one way to introduce biological information in order to improve the detection of differentially expressed genes and pathways. In this paper we show that the use of network models while still subject to the problems of normalization is a more robust method for detecting pathways that are differentially overrepresented in lung cancer data. Such differences may provide opportunities for novel therapeutics. In addition, we present evidence that non-small cell lung carcinoma is not a series of homogeneous diseases; rather that there is a heterogeny within the genotype which defies phenotype classification. This diversity helps to explain the lack of progress in developing therapies against non-small cell carcinoma and suggests that drug development may consider multiple pathways as treatment targets. </p>\",\"PeriodicalId\":56355,\"journal\":{\"name\":\"Microarrays\",\"volume\":\"3 4\",\"pages\":\"212-25\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2014-10-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.3390/microarrays3040212\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Microarrays\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/microarrays3040212\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microarrays","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/microarrays3040212","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
The Robustness of Pathway Analysis in Identifying Potential Drug Targets in Non-Small Cell Lung Carcinoma.
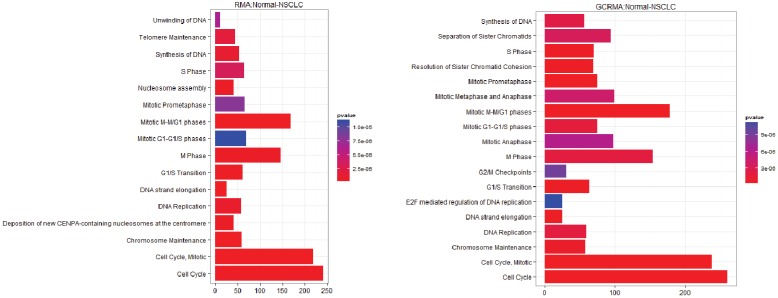
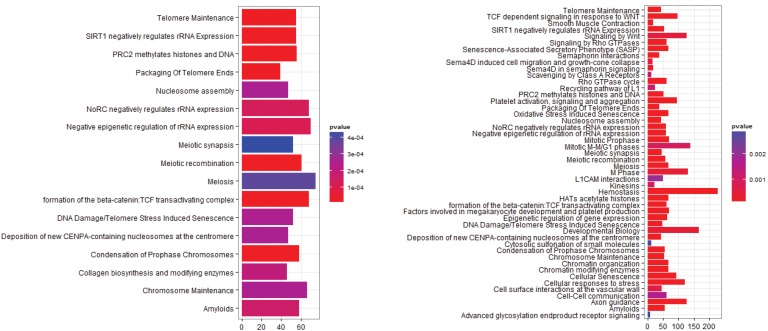
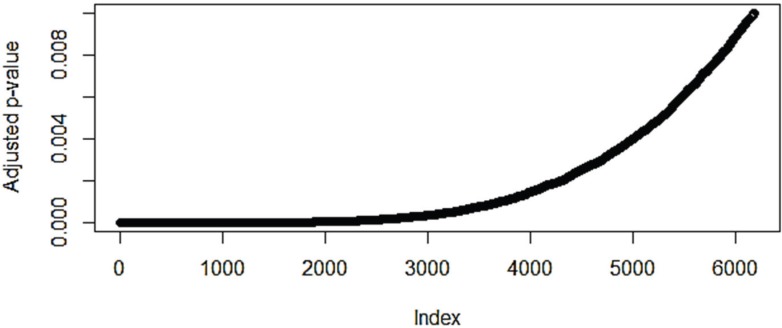
The identification of genes responsible for causing cancers from gene expression data has had varied success. Often the genes identified depend on the methods used for detecting expression patterns, or on the ways that the data had been normalized and filtered. The use of gene set enrichment analysis is one way to introduce biological information in order to improve the detection of differentially expressed genes and pathways. In this paper we show that the use of network models while still subject to the problems of normalization is a more robust method for detecting pathways that are differentially overrepresented in lung cancer data. Such differences may provide opportunities for novel therapeutics. In addition, we present evidence that non-small cell lung carcinoma is not a series of homogeneous diseases; rather that there is a heterogeny within the genotype which defies phenotype classification. This diversity helps to explain the lack of progress in developing therapies against non-small cell carcinoma and suggests that drug development may consider multiple pathways as treatment targets.
期刊介绍:
High-Throughput (formerly Microarrays, ISSN 2076-3905) is a multidisciplinary peer-reviewed scientific journal that provides an advanced forum for the publication of studies reporting high-dimensional approaches and developments in Life Sciences, Chemistry and related fields. Our aim is to encourage scientists to publish their experimental and theoretical results based on high-throughput techniques as well as computational and statistical tools for data analysis and interpretation. The full experimental or methodological details must be provided so that the results can be reproduced. There is no restriction on the length of the papers. High-Throughput invites submissions covering several topics, including, but not limited to: Microarrays, DNA Sequencing, RNA Sequencing, Protein Identification and Quantification, Cell-based Approaches, Omics Technologies, Imaging, Bioinformatics, Computational Biology/Chemistry, Statistics, Integrative Omics, Drug Discovery and Development, Microfluidics, Lab-on-a-chip, Data Mining, Databases, Multiplex Assays.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


