Ezequiel Luis Nicolazzi, Gabriele Marras, Alessandra Stella
{"title":"SNPConvert:家畜SNP阵列标准化与整合。","authors":"Ezequiel Luis Nicolazzi, Gabriele Marras, Alessandra Stella","doi":"10.3390/microarrays5020017","DOIUrl":null,"url":null,"abstract":"<p><p>One of the main advantages of single nucleotide polymorphism (SNP) array technology is providing genotype calls for a specific number of SNP markers at a relatively low cost. Since its first application in animal genetics, the number of available SNP arrays for each species has been constantly increasing. However, conversely to that observed in whole genome sequence data analysis, SNP array data does not have a common set of file formats or coding conventions for allele calling. Therefore, the standardization and integration of SNP array data from multiple sources have become an obstacle, especially for users with basic or no programming skills. Here, we describe the difficulties related to handling SNP array data, focusing on file formats, SNP allele coding, and mapping. We also present SNPConvert suite, a multi-platform, open-source, and user-friendly set of tools to overcome these issues. This tool, which can be integrated with open-source and open-access tools already available, is a first step towards an integrated system to standardize and integrate any type of raw SNP array data. The tool is available at: https://github. com/nicolazzie/SNPConvert.git. </p>","PeriodicalId":56355,"journal":{"name":"Microarrays","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2016-06-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.3390/microarrays5020017","citationCount":"7","resultStr":"{\"title\":\"SNPConvert: SNP Array Standardization and Integration in Livestock Species.\",\"authors\":\"Ezequiel Luis Nicolazzi, Gabriele Marras, Alessandra Stella\",\"doi\":\"10.3390/microarrays5020017\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>One of the main advantages of single nucleotide polymorphism (SNP) array technology is providing genotype calls for a specific number of SNP markers at a relatively low cost. Since its first application in animal genetics, the number of available SNP arrays for each species has been constantly increasing. However, conversely to that observed in whole genome sequence data analysis, SNP array data does not have a common set of file formats or coding conventions for allele calling. Therefore, the standardization and integration of SNP array data from multiple sources have become an obstacle, especially for users with basic or no programming skills. Here, we describe the difficulties related to handling SNP array data, focusing on file formats, SNP allele coding, and mapping. We also present SNPConvert suite, a multi-platform, open-source, and user-friendly set of tools to overcome these issues. This tool, which can be integrated with open-source and open-access tools already available, is a first step towards an integrated system to standardize and integrate any type of raw SNP array data. The tool is available at: https://github. com/nicolazzie/SNPConvert.git. </p>\",\"PeriodicalId\":56355,\"journal\":{\"name\":\"Microarrays\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2016-06-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.3390/microarrays5020017\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Microarrays\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/microarrays5020017\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microarrays","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/microarrays5020017","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
SNPConvert: SNP Array Standardization and Integration in Livestock Species.
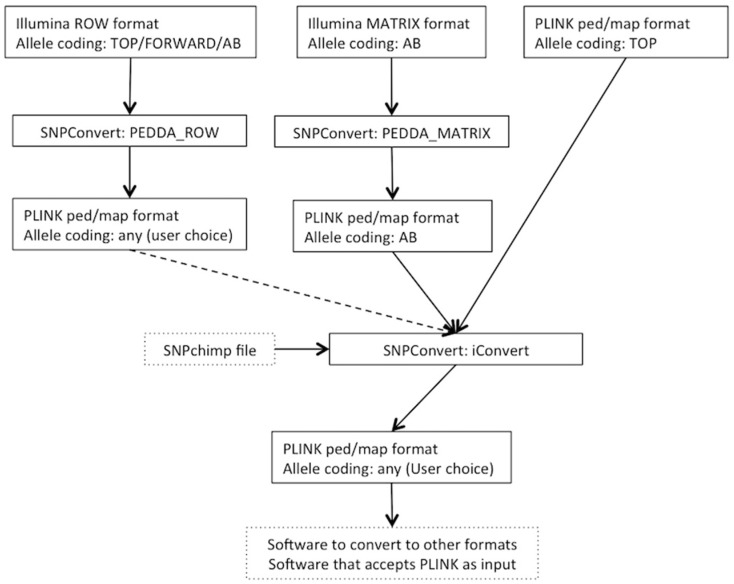
One of the main advantages of single nucleotide polymorphism (SNP) array technology is providing genotype calls for a specific number of SNP markers at a relatively low cost. Since its first application in animal genetics, the number of available SNP arrays for each species has been constantly increasing. However, conversely to that observed in whole genome sequence data analysis, SNP array data does not have a common set of file formats or coding conventions for allele calling. Therefore, the standardization and integration of SNP array data from multiple sources have become an obstacle, especially for users with basic or no programming skills. Here, we describe the difficulties related to handling SNP array data, focusing on file formats, SNP allele coding, and mapping. We also present SNPConvert suite, a multi-platform, open-source, and user-friendly set of tools to overcome these issues. This tool, which can be integrated with open-source and open-access tools already available, is a first step towards an integrated system to standardize and integrate any type of raw SNP array data. The tool is available at: https://github. com/nicolazzie/SNPConvert.git.
期刊介绍:
High-Throughput (formerly Microarrays, ISSN 2076-3905) is a multidisciplinary peer-reviewed scientific journal that provides an advanced forum for the publication of studies reporting high-dimensional approaches and developments in Life Sciences, Chemistry and related fields. Our aim is to encourage scientists to publish their experimental and theoretical results based on high-throughput techniques as well as computational and statistical tools for data analysis and interpretation. The full experimental or methodological details must be provided so that the results can be reproduced. There is no restriction on the length of the papers. High-Throughput invites submissions covering several topics, including, but not limited to: Microarrays, DNA Sequencing, RNA Sequencing, Protein Identification and Quantification, Cell-based Approaches, Omics Technologies, Imaging, Bioinformatics, Computational Biology/Chemistry, Statistics, Integrative Omics, Drug Discovery and Development, Microfluidics, Lab-on-a-chip, Data Mining, Databases, Multiplex Assays.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


