Agnese Viluma, Shumaila Sayyab, Sofia Mikko, Göran Andersson, Tomas F Bergström
{"title":"4只中华冠犬全基因组测序在离子质子系统变异检测中的评价。","authors":"Agnese Viluma, Shumaila Sayyab, Sofia Mikko, Göran Andersson, Tomas F Bergström","doi":"10.1186/s40575-015-0029-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Next generation sequencing (NGS) has traditionally been performed by large genome centers, but in recent years, the costs for whole-genome sequencing (WGS) have decreased substantially. With the introduction of smaller and less expensive \"desktop\" systems, NGS is now moving into the general laboratory. To evaluate the Ion Proton system for WGS we sequenced four Chinese Crested dogs and analyzed the data quality in terms of genome and exome coverage, the number of detected single nucleotide variants (SNVs) and insertions and deletions (INDELs) and the genotype concordance with the Illumina HD canine SNP array. For each of the four dogs, a 200 bp fragment library was constructed from genomic DNA and sequenced on two Ion PI chips per dog to reach mean coverage of 6-8x of the canine genome (genome size ≈ 2.4 Gb).</p><p><strong>Results: </strong>On average, each Ion PI chip yielded approximately 73.3 million reads with a mean read length of 130 bp (~9.5 Gb sequence data) of which 98.5 % could be aligned to the canine reference genome (CanFam3.1). By sequencing a single dog using one fragment library and two Ion PI chips, on average 80 % of the genome and 77 % exome was covered by at least four reads. After removing duplicate reads (20.7 %) the mean coverage across the whole genome was 6x. Using sequence data from all four individuals (four fragment libraries and eight Ion PI chips) the genome and exome coverage could be further increased to 97.2 and 94.3 %, respectively. We detected 4.83 million unique SNPs and 6.10 million unique INDEL positions across all individuals. A comparison between SNP genotypes detected with the WGS and the 170 K Illumina HD canine SNP array showed 90 % concordance.</p><p><strong>Conclusions: </strong>We have evaluated whole-genome sequencing on the Ion Proton system for genetic variant detection in four Chinese crested dogs. Even though INDEL calling with Ion Proton data is challenging due to specific platform errors, in case of SNP calling it can serve as an alternative to other next-generation sequencing platforms and SNP genotyping arrays, in studies aiming to identify causative mutations for rare monogenic diseases. In addition, we have identified new genetic variants of the Chinese Crested dog that will contribute to further whole-genome sequencing studies aimed to identify mutations associated with monogenic diseases with autosomal recessive inheritance.</p>","PeriodicalId":91060,"journal":{"name":"Canine genetics and epidemiology","volume":"2 ","pages":"16"},"PeriodicalIF":0.0000,"publicationDate":"2015-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s40575-015-0029-2","citationCount":"5","resultStr":"{\"title\":\"Evaluation of whole-genome sequencing of four Chinese crested dogs for variant detection using the ion proton system.\",\"authors\":\"Agnese Viluma, Shumaila Sayyab, Sofia Mikko, Göran Andersson, Tomas F Bergström\",\"doi\":\"10.1186/s40575-015-0029-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Next generation sequencing (NGS) has traditionally been performed by large genome centers, but in recent years, the costs for whole-genome sequencing (WGS) have decreased substantially. With the introduction of smaller and less expensive \\\"desktop\\\" systems, NGS is now moving into the general laboratory. To evaluate the Ion Proton system for WGS we sequenced four Chinese Crested dogs and analyzed the data quality in terms of genome and exome coverage, the number of detected single nucleotide variants (SNVs) and insertions and deletions (INDELs) and the genotype concordance with the Illumina HD canine SNP array. For each of the four dogs, a 200 bp fragment library was constructed from genomic DNA and sequenced on two Ion PI chips per dog to reach mean coverage of 6-8x of the canine genome (genome size ≈ 2.4 Gb).</p><p><strong>Results: </strong>On average, each Ion PI chip yielded approximately 73.3 million reads with a mean read length of 130 bp (~9.5 Gb sequence data) of which 98.5 % could be aligned to the canine reference genome (CanFam3.1). By sequencing a single dog using one fragment library and two Ion PI chips, on average 80 % of the genome and 77 % exome was covered by at least four reads. After removing duplicate reads (20.7 %) the mean coverage across the whole genome was 6x. Using sequence data from all four individuals (four fragment libraries and eight Ion PI chips) the genome and exome coverage could be further increased to 97.2 and 94.3 %, respectively. We detected 4.83 million unique SNPs and 6.10 million unique INDEL positions across all individuals. A comparison between SNP genotypes detected with the WGS and the 170 K Illumina HD canine SNP array showed 90 % concordance.</p><p><strong>Conclusions: </strong>We have evaluated whole-genome sequencing on the Ion Proton system for genetic variant detection in four Chinese crested dogs. Even though INDEL calling with Ion Proton data is challenging due to specific platform errors, in case of SNP calling it can serve as an alternative to other next-generation sequencing platforms and SNP genotyping arrays, in studies aiming to identify causative mutations for rare monogenic diseases. In addition, we have identified new genetic variants of the Chinese Crested dog that will contribute to further whole-genome sequencing studies aimed to identify mutations associated with monogenic diseases with autosomal recessive inheritance.</p>\",\"PeriodicalId\":91060,\"journal\":{\"name\":\"Canine genetics and epidemiology\",\"volume\":\"2 \",\"pages\":\"16\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2015-10-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s40575-015-0029-2\",\"citationCount\":\"5\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Canine genetics and epidemiology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s40575-015-0029-2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2015/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Canine genetics and epidemiology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40575-015-0029-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2015/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Evaluation of whole-genome sequencing of four Chinese crested dogs for variant detection using the ion proton system.
Background: Next generation sequencing (NGS) has traditionally been performed by large genome centers, but in recent years, the costs for whole-genome sequencing (WGS) have decreased substantially. With the introduction of smaller and less expensive "desktop" systems, NGS is now moving into the general laboratory. To evaluate the Ion Proton system for WGS we sequenced four Chinese Crested dogs and analyzed the data quality in terms of genome and exome coverage, the number of detected single nucleotide variants (SNVs) and insertions and deletions (INDELs) and the genotype concordance with the Illumina HD canine SNP array. For each of the four dogs, a 200 bp fragment library was constructed from genomic DNA and sequenced on two Ion PI chips per dog to reach mean coverage of 6-8x of the canine genome (genome size ≈ 2.4 Gb).
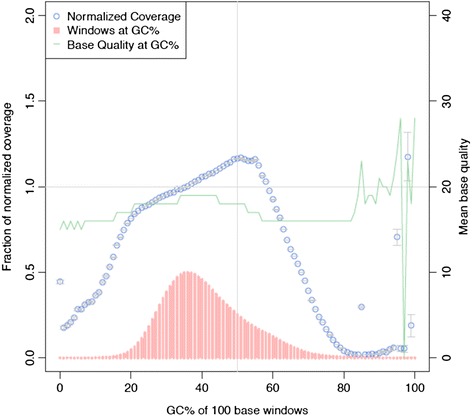
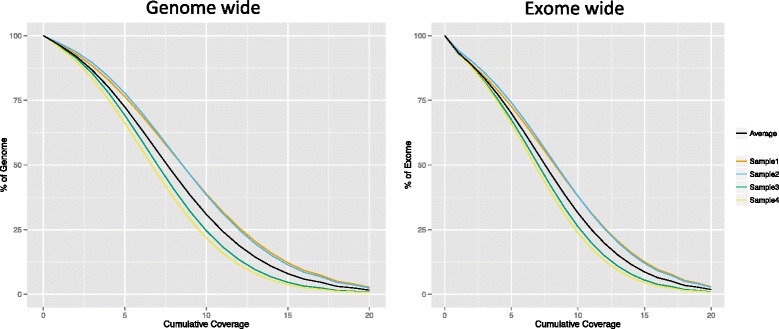
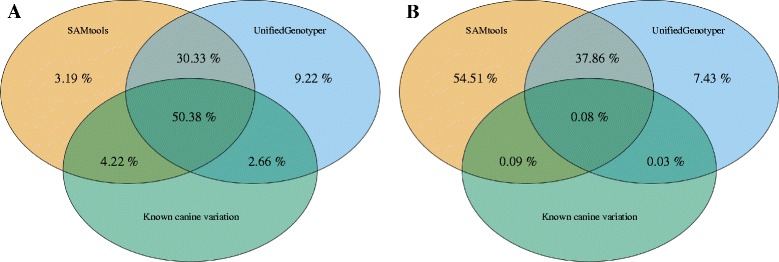
Results: On average, each Ion PI chip yielded approximately 73.3 million reads with a mean read length of 130 bp (~9.5 Gb sequence data) of which 98.5 % could be aligned to the canine reference genome (CanFam3.1). By sequencing a single dog using one fragment library and two Ion PI chips, on average 80 % of the genome and 77 % exome was covered by at least four reads. After removing duplicate reads (20.7 %) the mean coverage across the whole genome was 6x. Using sequence data from all four individuals (four fragment libraries and eight Ion PI chips) the genome and exome coverage could be further increased to 97.2 and 94.3 %, respectively. We detected 4.83 million unique SNPs and 6.10 million unique INDEL positions across all individuals. A comparison between SNP genotypes detected with the WGS and the 170 K Illumina HD canine SNP array showed 90 % concordance.
Conclusions: We have evaluated whole-genome sequencing on the Ion Proton system for genetic variant detection in four Chinese crested dogs. Even though INDEL calling with Ion Proton data is challenging due to specific platform errors, in case of SNP calling it can serve as an alternative to other next-generation sequencing platforms and SNP genotyping arrays, in studies aiming to identify causative mutations for rare monogenic diseases. In addition, we have identified new genetic variants of the Chinese Crested dog that will contribute to further whole-genome sequencing studies aimed to identify mutations associated with monogenic diseases with autosomal recessive inheritance.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


