S A Kocheva, M Gjorgjievska, K Martinova, Z Antevska-Trajkova, A Jovanovska, D Plaseska-Karanfilska
{"title":"先天性角化不良患者的新发TINF2 C.845G>A致病变异","authors":"S A Kocheva, M Gjorgjievska, K Martinova, Z Antevska-Trajkova, A Jovanovska, D Plaseska-Karanfilska","doi":"10.2478/bjmg-2021-0027","DOIUrl":null,"url":null,"abstract":"<p><p>Dyskeratosis congenita (DC) is a clinically and genetically heterogeneous, multisystem inherited syndrome with a very high risk for bone marrow failure (BMF) and cancer predisposition. The classical clinical form of DC is characterized by abnormal skin pigmentation, nail dystrophy, and oral leukoplakia. Bone marrow failure is considered to be an important and major complication of DC and the leading cause of death which develops in around 85% of cases. A number of genes involved in telomere maintenance are associated with DC, such as genes that encode the components of the telomerase complex (<i>TERT</i>, <i>DKC1</i>, <i>TERC</i>, <i>NOP</i>10, and <i>NHP</i>2), T-loop assembly protein (<i>RTEL1</i>), telomere capping (<i>CTC1</i>), telomere shelterin complex (<i>TINF2</i>), and telomerase trafficking protein (<i>TCAB1</i>). Mutations in <i>TINF2</i> have been reported in 11-20% of all patients with DC and have been associated with bone marrow failure. Here we report on a 19-month old boy with very early presentation of bone marrow failure as a first clinical manifestation of DC. Upon first admission, the patient presented with thrombocytopenia and macrocytic anemia. Soon after, his blood counts deteriorated with the development of pancytopenia and aplastic anemia. Four months later, he developed nail dystrophy and skin hyperpigmentation. A <i>de novo</i> heterozygous pathogenic variant c.845G>A, p.(Arg282His) was located in exon 6 of <i>TINF2</i> gene and was identified via clinical exome sequencing. The findings confirmed the diagnosis of DC. This is the first case with DC due to <i>TINF2</i> pathogenic variant reported in North Macedonia.</p>","PeriodicalId":55403,"journal":{"name":"Balkan Journal of Medical Genetics","volume":"24 2","pages":"89-93"},"PeriodicalIF":0.9000,"publicationDate":"2022-06-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/9f/fa/bjmg-24-089.PMC9524180.pdf","citationCount":"0","resultStr":"{\"title\":\"<i>de Novo TINF2</i> C.845G>A: Pathogenic Variant in Patient with Dyskeratosis Congenita.\",\"authors\":\"S A Kocheva, M Gjorgjievska, K Martinova, Z Antevska-Trajkova, A Jovanovska, D Plaseska-Karanfilska\",\"doi\":\"10.2478/bjmg-2021-0027\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Dyskeratosis congenita (DC) is a clinically and genetically heterogeneous, multisystem inherited syndrome with a very high risk for bone marrow failure (BMF) and cancer predisposition. The classical clinical form of DC is characterized by abnormal skin pigmentation, nail dystrophy, and oral leukoplakia. Bone marrow failure is considered to be an important and major complication of DC and the leading cause of death which develops in around 85% of cases. A number of genes involved in telomere maintenance are associated with DC, such as genes that encode the components of the telomerase complex (<i>TERT</i>, <i>DKC1</i>, <i>TERC</i>, <i>NOP</i>10, and <i>NHP</i>2), T-loop assembly protein (<i>RTEL1</i>), telomere capping (<i>CTC1</i>), telomere shelterin complex (<i>TINF2</i>), and telomerase trafficking protein (<i>TCAB1</i>). Mutations in <i>TINF2</i> have been reported in 11-20% of all patients with DC and have been associated with bone marrow failure. Here we report on a 19-month old boy with very early presentation of bone marrow failure as a first clinical manifestation of DC. Upon first admission, the patient presented with thrombocytopenia and macrocytic anemia. Soon after, his blood counts deteriorated with the development of pancytopenia and aplastic anemia. Four months later, he developed nail dystrophy and skin hyperpigmentation. A <i>de novo</i> heterozygous pathogenic variant c.845G>A, p.(Arg282His) was located in exon 6 of <i>TINF2</i> gene and was identified via clinical exome sequencing. The findings confirmed the diagnosis of DC. This is the first case with DC due to <i>TINF2</i> pathogenic variant reported in North Macedonia.</p>\",\"PeriodicalId\":55403,\"journal\":{\"name\":\"Balkan Journal of Medical Genetics\",\"volume\":\"24 2\",\"pages\":\"89-93\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2022-06-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/9f/fa/bjmg-24-089.PMC9524180.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Balkan Journal of Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.2478/bjmg-2021-0027\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/11/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Balkan Journal of Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2478/bjmg-2021-0027","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/11/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
de Novo TINF2 C.845G>A: Pathogenic Variant in Patient with Dyskeratosis Congenita.
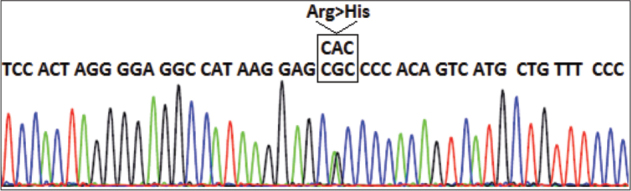
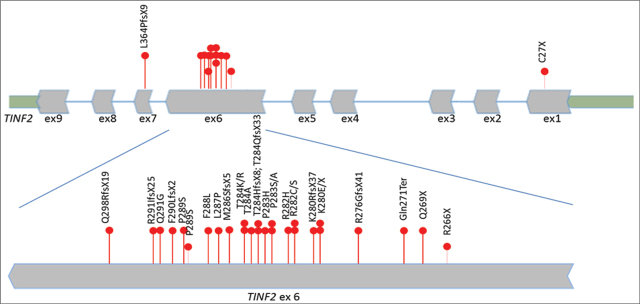
Dyskeratosis congenita (DC) is a clinically and genetically heterogeneous, multisystem inherited syndrome with a very high risk for bone marrow failure (BMF) and cancer predisposition. The classical clinical form of DC is characterized by abnormal skin pigmentation, nail dystrophy, and oral leukoplakia. Bone marrow failure is considered to be an important and major complication of DC and the leading cause of death which develops in around 85% of cases. A number of genes involved in telomere maintenance are associated with DC, such as genes that encode the components of the telomerase complex (TERT, DKC1, TERC, NOP10, and NHP2), T-loop assembly protein (RTEL1), telomere capping (CTC1), telomere shelterin complex (TINF2), and telomerase trafficking protein (TCAB1). Mutations in TINF2 have been reported in 11-20% of all patients with DC and have been associated with bone marrow failure. Here we report on a 19-month old boy with very early presentation of bone marrow failure as a first clinical manifestation of DC. Upon first admission, the patient presented with thrombocytopenia and macrocytic anemia. Soon after, his blood counts deteriorated with the development of pancytopenia and aplastic anemia. Four months later, he developed nail dystrophy and skin hyperpigmentation. A de novo heterozygous pathogenic variant c.845G>A, p.(Arg282His) was located in exon 6 of TINF2 gene and was identified via clinical exome sequencing. The findings confirmed the diagnosis of DC. This is the first case with DC due to TINF2 pathogenic variant reported in North Macedonia.
期刊介绍:
Balkan Journal of Medical Genetics is a journal in the English language for publication of articles involving all branches of medical genetics: human cytogenetics, molecular genetics, clinical genetics, immunogenetics, oncogenetics, pharmacogenetics, population genetics, genetic screening and diagnosis of monogenic and polygenic diseases, prenatal and preimplantation genetic diagnosis, genetic counselling, advances in treatment and prevention.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


