{"title":"抑制MAD2L1通过线粒体功能损伤和诱导细胞衰老介导肺纤维化。","authors":"Lan Wang, Ruyan Wan, Xinyu Chen, Xiaoshu Guo, Zhongzheng Li, Weiming Zhao, Peishuo Yan, Guoying Yu","doi":"10.1155/2022/9663354","DOIUrl":null,"url":null,"abstract":"<p><p>Idiopathic pulmonary fibrosis (IPF) is a chronic, irreversible, and progressive interstitial lung disease characterized by recurrent alveolar epithelial cell injury, fibroblast hyperproliferation, and cumulative deposition of extracellular matrix leading to alveolar destruction in the lungs. Mitotic arrest deficient 2 like 1 (MAD2L1) is a component of the mitotic spindle assembly checkpoint that prevents the onset of anaphase until all chromosomes are properly aligned at metaphase and is a potential therapeutic target in cancers. However, the role of MAD2L1 in pulmonary fibrosis has not been explored. We analyzed the expression of MAD2L1 in lung tissues from control subjects, IPF patients, and mice with bleomycin-induced fibrosis via IHC, qRT-PCR, and Western blot analysis. We examined the roles of MAD2L1 in ROS production, mitochondrial function, cell senescence, and the establishment of a profibrotic microenvironment. We found that MAD2L1 was highly upregulated in alveolar epithelial cells in fibrotic lung tissues from both patients with IPF and mice with bleomycin-induced fibrosis. Loss of MAD2L1 expression or activity led to decreases of cell viability and proliferation in A549 cells. Subsequent mechanistic investigation demonstrated that inhibition of MAD2L1 damaged mitochondria, which led to augmented ROS production and cellular senescence, and thus promoted the establishment of a profibrotic microenvironment. Taken together, these results reveal that alleviation of alveolar epithelial cell mitochondrial damage arising from augmentation of MAD2L1 may be a novel therapeutic strategy for mitigating pulmonary fibrosis.</p>","PeriodicalId":9416,"journal":{"name":"Canadian respiratory journal","volume":" ","pages":"9663354"},"PeriodicalIF":2.1000,"publicationDate":"2022-10-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9553670/pdf/","citationCount":"1","resultStr":"{\"title\":\"Inhibition of MAD2L1 Mediates Pulmonary Fibrosis through Impairment of Mitochondrial Function and Induction of Cell Senescence.\",\"authors\":\"Lan Wang, Ruyan Wan, Xinyu Chen, Xiaoshu Guo, Zhongzheng Li, Weiming Zhao, Peishuo Yan, Guoying Yu\",\"doi\":\"10.1155/2022/9663354\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Idiopathic pulmonary fibrosis (IPF) is a chronic, irreversible, and progressive interstitial lung disease characterized by recurrent alveolar epithelial cell injury, fibroblast hyperproliferation, and cumulative deposition of extracellular matrix leading to alveolar destruction in the lungs. Mitotic arrest deficient 2 like 1 (MAD2L1) is a component of the mitotic spindle assembly checkpoint that prevents the onset of anaphase until all chromosomes are properly aligned at metaphase and is a potential therapeutic target in cancers. However, the role of MAD2L1 in pulmonary fibrosis has not been explored. We analyzed the expression of MAD2L1 in lung tissues from control subjects, IPF patients, and mice with bleomycin-induced fibrosis via IHC, qRT-PCR, and Western blot analysis. We examined the roles of MAD2L1 in ROS production, mitochondrial function, cell senescence, and the establishment of a profibrotic microenvironment. We found that MAD2L1 was highly upregulated in alveolar epithelial cells in fibrotic lung tissues from both patients with IPF and mice with bleomycin-induced fibrosis. Loss of MAD2L1 expression or activity led to decreases of cell viability and proliferation in A549 cells. Subsequent mechanistic investigation demonstrated that inhibition of MAD2L1 damaged mitochondria, which led to augmented ROS production and cellular senescence, and thus promoted the establishment of a profibrotic microenvironment. Taken together, these results reveal that alleviation of alveolar epithelial cell mitochondrial damage arising from augmentation of MAD2L1 may be a novel therapeutic strategy for mitigating pulmonary fibrosis.</p>\",\"PeriodicalId\":9416,\"journal\":{\"name\":\"Canadian respiratory journal\",\"volume\":\" \",\"pages\":\"9663354\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2022-10-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9553670/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Canadian respiratory journal\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1155/2022/9663354\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"RESPIRATORY SYSTEM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Canadian respiratory journal","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1155/2022/9663354","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"RESPIRATORY SYSTEM","Score":null,"Total":0}
Inhibition of MAD2L1 Mediates Pulmonary Fibrosis through Impairment of Mitochondrial Function and Induction of Cell Senescence.
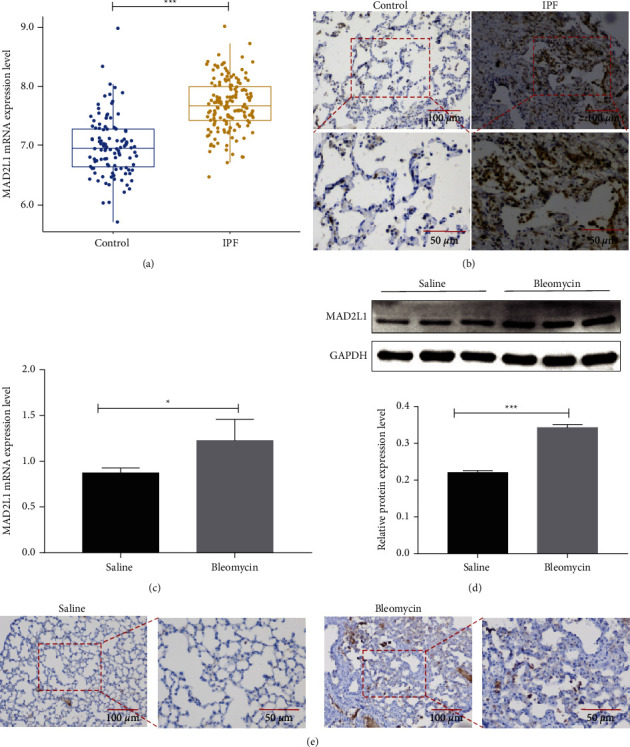
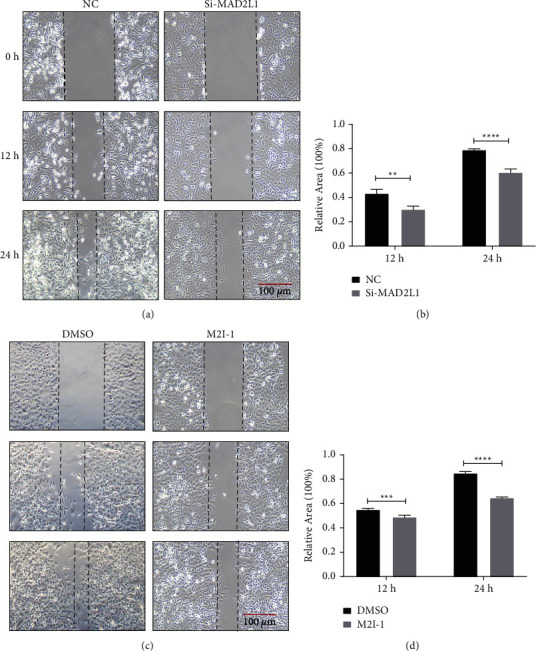
Idiopathic pulmonary fibrosis (IPF) is a chronic, irreversible, and progressive interstitial lung disease characterized by recurrent alveolar epithelial cell injury, fibroblast hyperproliferation, and cumulative deposition of extracellular matrix leading to alveolar destruction in the lungs. Mitotic arrest deficient 2 like 1 (MAD2L1) is a component of the mitotic spindle assembly checkpoint that prevents the onset of anaphase until all chromosomes are properly aligned at metaphase and is a potential therapeutic target in cancers. However, the role of MAD2L1 in pulmonary fibrosis has not been explored. We analyzed the expression of MAD2L1 in lung tissues from control subjects, IPF patients, and mice with bleomycin-induced fibrosis via IHC, qRT-PCR, and Western blot analysis. We examined the roles of MAD2L1 in ROS production, mitochondrial function, cell senescence, and the establishment of a profibrotic microenvironment. We found that MAD2L1 was highly upregulated in alveolar epithelial cells in fibrotic lung tissues from both patients with IPF and mice with bleomycin-induced fibrosis. Loss of MAD2L1 expression or activity led to decreases of cell viability and proliferation in A549 cells. Subsequent mechanistic investigation demonstrated that inhibition of MAD2L1 damaged mitochondria, which led to augmented ROS production and cellular senescence, and thus promoted the establishment of a profibrotic microenvironment. Taken together, these results reveal that alleviation of alveolar epithelial cell mitochondrial damage arising from augmentation of MAD2L1 may be a novel therapeutic strategy for mitigating pulmonary fibrosis.
期刊介绍:
Canadian Respiratory Journal is a peer-reviewed, Open Access journal that aims to provide a multidisciplinary forum for research in all areas of respiratory medicine. The journal publishes original research articles, review articles, and clinical studies related to asthma, allergy, COPD, non-invasive ventilation, therapeutic intervention, lung cancer, airway and lung infections, as well as any other respiratory diseases.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


